Protracted Withdrawal is Sensitivity To the Sulfite Paradox and Modern-day Lions
© 2025 by Meredith Arthur, MS, RD, LD is licensed under CC BY-NC-SA 4.0
Last night, Jan’s husband held off washing the dishes after dinner because Jan wanted a super hot bath as a treat after a long day of caring for her rambunctious children. Jan suffers from extreme premenstrual syndrome and has been diagnosed with premenstrual dysphoric disorder (PMMD), which makes her easily irritated with her children before the onset of her period. She was prescribed selective serotonin reuptake inhibitors (SSRI) and Xanax, a benzodiazepine, to help with her anxiety and mood swings during high school and continued them through college. After graduation and getting married, she decided to go holistic in preparation for having children, so she weaned off the pharmaceuticals but experienced severe withdrawal symptoms. She and her husband, Dave, had three lovely children in the past five years after she successfully got off of benzos and recovered from withdrawal. Those scary days haunted her, and she always worried about relapsing into the nightmare. Now, she naturally treats her PMMD, including soothing baths to ease tension. Her baths were usually hot but not as hot as she wanted, and her husband knew that and knew she truly needed something to calm her very on-edge body that night.
Jan felt a bit dizzy after the bath but headed off to bed immediately and fell fast asleep until around 2 AM when, to her horror, her heartbeat woke her up. The familiar pounding sensation couldn’t be real! How is this possible? She sat up in bed and immediately felt nauseous. She was going to vomit. She stood up and realized her feet were on fire, and her muscles were so tight that each step felt like stabbing pains. As she stumbled to the bathroom, her anxiety increased, and she felt the air hunger hit hard. She screamed in horror, “No. It’s back. I’m not okay. I’m not okay. I’m not okay,” while she sank to the floor next to the toilet. Dave, startled from sleep, ran into the bathroom and squatted next to her. He was scared to touch her because he knew she might not be able to handle his touch now. He had spent years waiting to be able to feel her soft skin with his hands, and tears streamed down his face as he realized it would be a long time before her body felt safe enough for a hug. They both stared at each other in terror. How could this be possible? Life was so good.
***********************************************************************************************************Often, in the protracted withdrawal community, people are told, “Only time can heal.” Well, if a moment in time like the one above can undo healing, how is time the only thing that can heal? ***********************************************************************************************************
Time does heal the brain. It takes time to restore GABA-A synapses, but why does a moment in time like the one above seemingly undo all the healing, causing setbacks sometimes to the point of being in acute withdrawal?
One metabolite that all humans share in varying degrees that can damage GABA-A synapses when made in excess in the human body is a derivative of sulfite, s-sulfocysteine (SSC). What if the quantity of s-sulfocysteine and sulfite determines your ability to stay healed in your body at any given moment? What if these moments in time that undo all the healing that time provides is an uptick in a naturally occurring compound that we all make every day? What if finding and avoiding triggers that increase this compound and getting better at sulfite metabolism is the key to getting and staying healed?

“Knowledge is the antidote to fear,- Knowledge, Use and Reason, with its higher aids. – Ralph Waldo Emerson
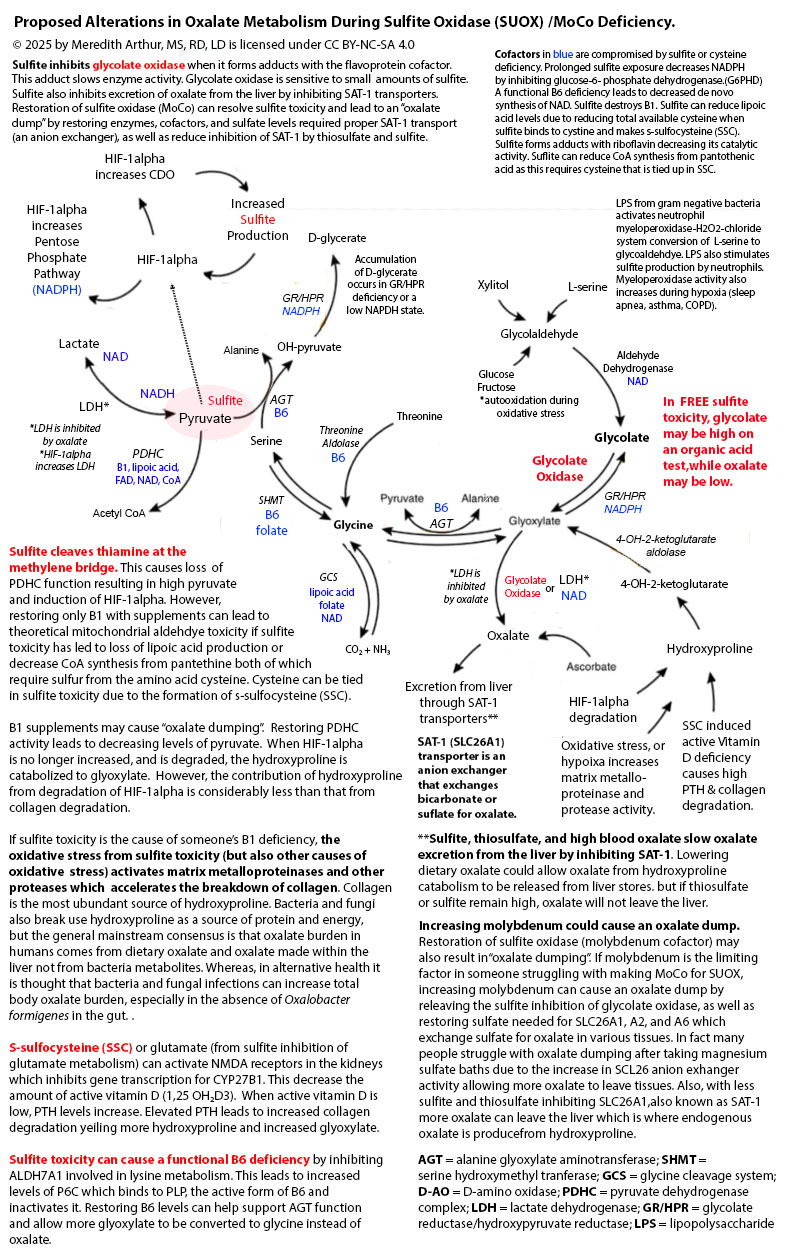
Meredith Arthur, MS, RD, LD believes that protracted withdrawal is an increased sensitivity to the Moco Steal and Sulfite Paradox, described by herself and Jenny Jones, PhD. This paradox is caused by an interplay between an immune and hypoxia pathway called hypoxia-inducible factor one alpha (HIF-1alpha) and a pathway called transsulfuration, where hydrogen sulfide, glutathione, taurine, thiosulfate, and sulfate are made. The triggering of the HIF-1alpha pathway leads to an increase in an enzyme called cysteine dioxygenase (CDO), leading to an overall rise in sulfite production, which must be metabolized to sulfate. Sulfate plays beneficial roles in the body, such as making healthy collagen, connective tissue, and myelin (a coating on nerves that makes them run smoothly), as well as being used for detoxifying toxins and hormone metabolism.
Sulfite, however, is destructive to the body. To deal with excess sulfite that isn’t made into sulfate, the body will bind it to cystine (two cysteine amino acids bound together) to produce s-sulfocysteine (SSC). Everyone makes SSC daily in varying amounts. Chris Masterjohn, PhD, provides an excellent summary of the variations in SSC among humans, and says, “You would think something that is present in everyone’s urine and varies 50-fold that happens to activate NMDA receptors and cause neurological degeneration could be a powerful explanation for a major part of the variability in trait anxiety, muscle tension, ease of being startled, and the risk of neurological and psychiatric disorders.” Meredith and Jenny agree with Chris’s astute observation and believe that individuals in their Facebook support group, The MoCo Steal: Escaping From the Sulfite Paradox, tend to be more sensitive to the damaging effects of SSC, but also sulfite itself. They believe that our members who have protracted withdrawal are struggling with elevated levels of SSC and sulfite toxicity.
Why, then, would humans even make this damaging version of sulfite? Meredith believes that during a period of increased need for the HIF-1alpha pathway, SSC can serve as an activator of NMDA receptors found in the brain, causing mental alertness and alterations in the metabolism of other organs throughout the body that have NMDA receptors. These alterations are required during a period of metabolic or immune stress. The issue arises when HIF-1alpha remains on for too long, leading to prolonged increases in CDO and sulfite, as seen in the diagram below.

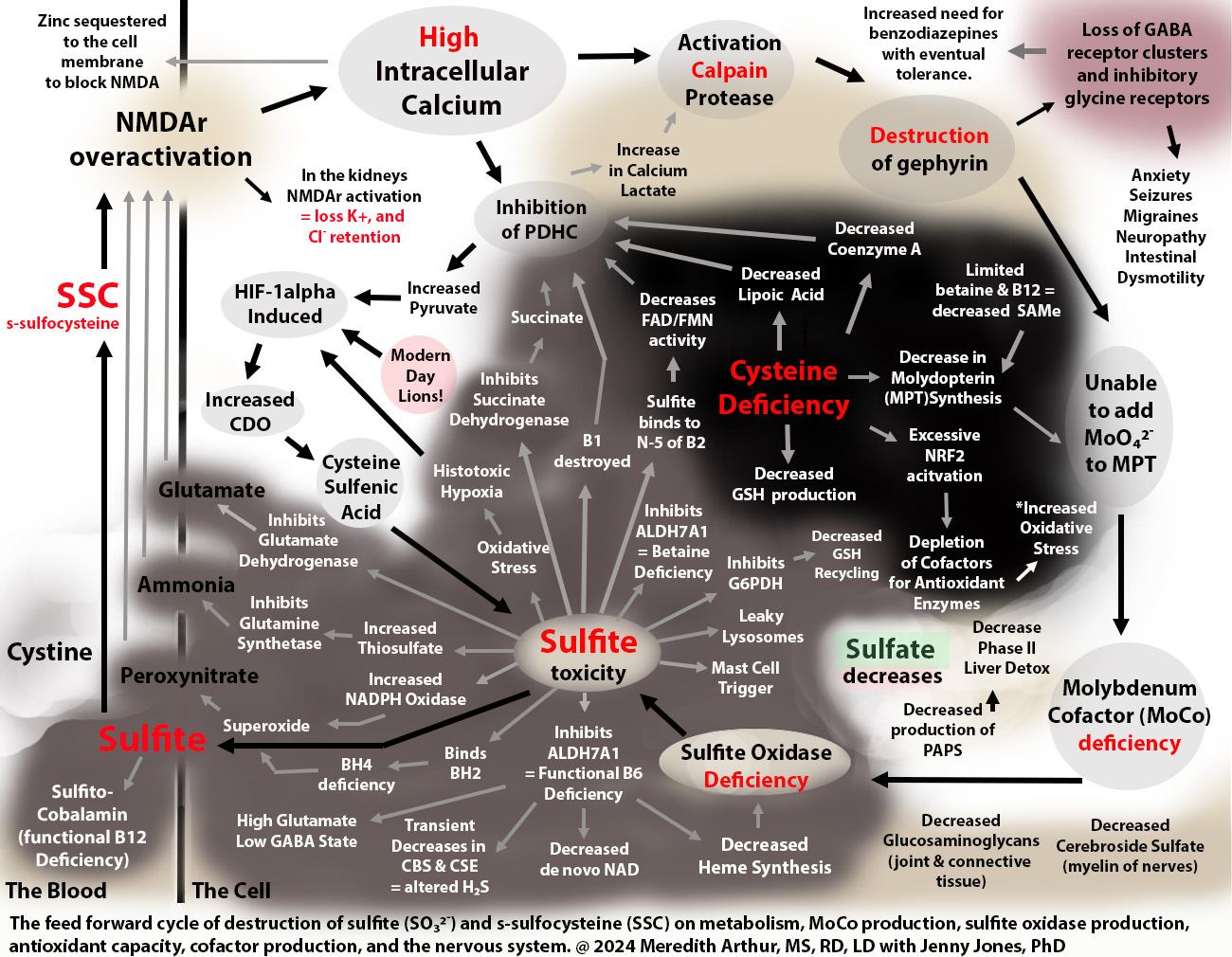
When excess sulfite accumulates, it can inhibit glutamate dehydrogenase activity, leading to an accumulation of glutamate and nerve damage. At the same time, sulfite can cause a functional B6 deficiency that causes a loss of the conversion of glutamate to GABA. GABA calms down nerve excitation. Even worse, as shown in the upper right-hand corner of the diagram above, excess SSC leads to a loss of GABAergic synapses. SSC or other metabolites over-activate NMDA receptors in the brain, leading to an excess amount of calcium entering cells. This activates calpain protease, which breaks down gephyrin, a glue that holds the GABA-A receptors in place. Loss of gephyrin leads to a high excitatory state in the brain due to the excessive activation of NMDA receptors without an adequate inhibitory response from GABA-A synapses. This loss of the glue, gephyrin, that holds GABA-A synapses in place is a contributor to insomnia, anxiety, nerve pain, and seizure disorder.
In addition, sulfite inhibits the process of methionine salvage by binding to B12 as well as by inhibiting the production of betaine, both of which are needed for restoring methionine levels to provide s-adenosyl methionine (SAM) required to make the molybdenum cofactor that is necessary for the enzyme sulfite oxidase. SSC, again, destroys gephyrin. Gephyrin isn’t just a glue that holds GABA receptors in place, but it is also the glue that sticks molybdenum into molydopterin to make MoCo. Without gephyrin, we can’t incorporate molybdenum into MoCo, no matter how much molybeneum is consumed. Thus, once out of control, sulfite prevents its metabolism by destroying the ability to make the cofactor for sulfite oxidase, MoCo, needed for its metabolism.
SSC Contributes to Tolerance
Individuals with high SSC levels feel anxious and have muscle spasms, insomnia, and sometimes seizures. They will often be prescribed benzodiazepines, which bind to what little GABA-A receptors are left and can calm down the excitatory actions of SSC. However, at some point, due to excessive activation of NMDA receptors and loss of gephyrin, the GABA-A receptors are no longer present in large enough quantities for any benzodiazepines to have a therapeutic effect. It is at this point that Meredith thinks that individuals on benzodiazepines experience “tolerance” and are weaned off of the drug as it is no longer working.
S-Sulfocysteine Helps Us Escape A Lion
One way to think of SSC is that it is a chemical that helps us escape a lion. Of course, none of us are escaping actual lions right now. However, modern-day lions induce the HIF-1alpha pathway, as depicted in the diagram below on the left. Any of these lions can lead to the overproduction of SSC and sulfite with the downstream damaging effects on health and metabolism shown in the diagram.

Even stressful moments, such as the loss of a loved one, the loss of a job, a divorce, or a massive unexpected bill, can lead to increases in adrenaline and norepinephrine that encourage unhealthy gut bacteria to grow. These unhealthy bacteria lead to bacterial toxins and leaky gut that induce the HIF-1alpha pathway and CDO and sulfite production. So, even that stressful moment in life can lead to an uptick in sulfite and SSC production and anxiety.
Jan, in the introduction, experienced low blood pressure due to a very hot bath while experiencing PMMD. Right before a woman’s period, the body has a surge of estrogen, which induces the HIF-1alpha pathway, but low blood pressure also induces the HIF-1alpha pathway. This leads to increased expression of CDO, resulting in more sulfite and SSC, leading to symptoms of protracted withdrawal. Often, individuals with protracted withdrawal are unable to pinpoint the exact cause of their regression into torment, and this may be due to HIF-1alpha being induced by many different unforeseen alterations in metabolism and sometimes circumstances beyond a person’s control. However, Dr. Jones and Meredith are working together to identify HIF-1alpha triggers, of which many are listed in the diagram above, to mitigate the uptick in this pathway in individuals vulnerable to sulfite and SSC toxicity.
Too much SSC because HIF-1alpha is stuck in the “on” mode!
Meredith believes that HIF-1alpha is stuck in an “on” mode for too long, leading to excess SSC production, but in the past, it was beneficial when truly running from danger. For the sake of simplicity, let’s use low oxygen and running from a lion as an example. While running from a lion, oxygen is used up quickly. When oxygen goes low in a cell, it induces the HIF-1alpha pathway, which increases CDO activity. This results in more production of sulfite and more SSC. SSC then fires up NMDA receptors in the central and peripheral nervous systems, which gives a sense of alertness, allowing us to identify if any more lions are hiding in the bushes ahead of us. This HIF-1alpha pathway also increases the total amount of blood vessels in the muscles so that the next time the lion attacks, more oxygen will be available to those tissues so that if the escape ever turns into a long-distance sprint, our muscles will be ready.
HIF-1alpha increases CDO activity, but it also changes metabolism so that a person makes energy quickly from glucose, which comes from carbohydrates (bread, cereal, rice, pasta, fruit, and vegetables). It also helps send glucose through a pathway called the pentose phosphate shunt that makes NADPH. NADPH helps recycle glutathione, a master antioxidant, from its used-up state (oxidized) to its fresh state (reduced). This reduced glutathione can mop up the oxidative stress that is happening. This change in metabolism can happen in any cell, including immune cells, to give them energy to fight infections. Unfortunately, excess sulfite can inhibit the enzyme that starts the pentose phosphate shunt, glucose-6-phosphate dehydrogenase, causing low levels of NADPH. Sulfite can also bind to oxidized glutathione, making recycling it back to reduced glutathione impossible.
Excess sulfite, however, should be metabolized to sulfate, not remain as sulfite. If done well, the intersection of HIF-1alpha and the transsulfuration pathway helps to increase the total amount of sulfate available in the body. This is if the enzyme that helps to make sulfate, sulfate oxidase (SUOX), is working well. SUOX is the end-game enzyme needed to prevent a feed-forward cycle that keeps the HIF-1alpha pathway turned on. This is because both SSC and sulfite will induce the HIF-1alpha pathway, which is explained in more detail in the summary of the MoCo Steal Leads to a Sulfite Paradox. Unfortunately, someone with acquired or genetic SUOX deficiency doesn’t make sulfate in this pathway. Instead, they have a build-up of sulfite, thiosulfate, and SSC.
How Benzodiazepines Lead to Overactivation of HIF-1alpha and a Moco Steal
MoCo is a cofactor made with Molybdenum (Mo), a mineral that is used only for five enzymes in the human body: xanthine oxidase (XO), xanthine dehydrogenase (XDH), aldehyde oxidase (AOX), sulfite oxidase (SUOX), and mitochondrial amidoxime reducing compound (mARC). All of these enzymes are necessary for metabolism, but SUOX is crucial to prevent sulfite toxicity. Please note that SUOX is the only enzyme that also requires heme for production. Heme synthesis is compromised in sulfite toxicity due to sulfite causing a functional B6 deficiency. This leads to the loss of pyridoxal-5-phosphate (P5P) needed for the first step in the heme synthesis pathway. Sulfite toxicity also causes the loss of lipoic acid, zinc, and copper needed for further steps in the heme pathway.

Jenny Jones, PhD, has developed a theory on how she suffered from and acquired sulfite oxidase deficiency, resulting in sulfite toxicity and excessive levels of SSC, glutamate, and peroxynitrate, which lead to adult onset seizure disorder. Her theory, The MoCol Steal, is that due to blocks in NAD production and recycling, and various other blocks at aldehyde dehydrogenase (ALDH), she had to use more of the low affinity, high-capacity enzyme for vitamin A metabolism, aldehyde oxidase (AOX) as well as XO and XDH that also participate in vitamin A metabolism during a low NAD state, the cofactor needed for alcohol dehydrogenase and ALDH. This caused a stealing of molybdenum cofactor towards the XOR family (shown on the left side of the diagram below) and away from sulfite oxidase (SO or SUOX, on the right side of the diagram below), leading to a loss of sulfite oxidase activity and her downward health spiral, from which she has now recovered.
Benzodiazepines can lead to Dr. Jones’s MoCo steal through alteration of immune function, resulting in an increased need for XO activity. In the gut, there are immune cells found in the gut-associated lymphoid tissue, including macrophages. Macrophages are like the beat cops of the immune system. They look out for pathogenic bacteria and viruses. Benzodiazepines alter macrophages by interacting with alpha-1 subunits on GABA-A receptors, leading to macrophages having acidic cytoplasms. This results in a decreased ability of these white blood cells to engulf and kill bacteria as well as to produce cytokines to attract other immune cells to the fight. Benzos can increase the risk for bacterial superinfections due to these alterations in macrophage activity. Benzos have also been shown to increase haemophilus parainfluenzae, a gram-negative bacteria that contains LPS and also produces H2S, which is metabolized to sulfite in the body and increases the need for molybdenum cofactor.
The immunosuppressive effects of benzodiazepines have been of great concern in the world of critical care as these are often used for sedation in intensive care units and have led to serious infections. Even so, while macrophages are incapacitated by benzos, other immune cells, called dendritic cells, are still available to recognize pathogens through toll-like receptors. This activates a pathway called NF-KB, which turns on an enzyme that makes hydrogen peroxide as a way to fight bacteria. This enzyme, xanthine oxidase, requires MoCo for activity, which leads to Dr. Jones’s Moco Steal. Dendritic cells also produce two more cytokines, IL-1 and TNF-1alph, which also increase xanthine oxidase levels. As the bacteria damage intestinal tissues, the infected gut tissue becomes hypoxic. Hypoxia induces the HIF-1apha pathway, leading to increased CDO activity and more sulfite, but the excess need for MoCo for xanthine oxidase has shifted molybdenum away from sulfite oxidase. Thus, benzodiazepines can lead to altered immune function that leads to Jenny Jones’s MoCo Steal, explained in the diagram above.
SSRI Alter Gut Microbiota.
Many individuals struggling with modern-day lions get symptom relief from benzodiazepines but often are prescribed serotonin reuptake inhibitors as well for symptoms of depression. There is an ongoing debate whether the serotonin deficiency theory of depression is valid, but the fact that SSRI can speed up or slow down intestinal movements has been confirmed. At minimum, we know that altering GI transit can alter the microbiome and SSRI have been found to alter the gut microbiota. In addition, similar to benzodiazepines, at some point individuals reach a tolerance level with SSRI. Weaning off of them, however, comes with withdrawal type symptoms and ongoing nuerologocial sequalae.
If Protracted Withdrawal is a Hypersensitivity to SSC and Sulfite toxicity, What Can I do?
The first step for dealing with an uptick in sulfite and SSC is to utilize what Jenny and Meredith call “mopper uppers” and modulators of NMDA receptors. Mopper uppers are nutrients that are capable of binding to sulfite in the body. Because they bind to sulfite, these mopper uppers are usually deficient in the body due to being bound to sulfite. Meredith especially recommends for periods of times that she calls, “I’m not okay, I’m not okay,” that you consider this regimen with your healthcare provider to immediately mop up sulfite. If you also have insomnia, which is attributed to the overactivation of NMDA receptors, she recommends this helpful sheet to you and your healthcare provider on how to slow down NMDA receptor activity. Finally, you can be aware of the “lions” found in the diagram early in this document as well as join the MoCo Steal: Recovering From Sulfite Toxicity Support Group to share what your “lions” are and contribute to the ongoing research on how to promote the healing process. Jenny and Meredith believe that identifying and avoiding lions is a key component to healing.
Do We Ever Fully Recover From the Sulfite Paradox/Protracted Withdrawal?
Meredith and Jenny are hopeful that now that the problem has been properly identified, we all can come to a healed state and that time is not the only healer. Unfortunately, the crossroads of HIF-1alpha and transsulfuration is never going away, but navigating the intersection will become easier, and preventing collisions, what those with benzo protracted withdrawal call “waves”, is possible. It’s not just time that heals, but it is time to take the fear out of protracted withdrawal. Find the lions and cage them. Mop up the sulfite. Restore lost nutrients. Slow the NMDA receptors down. Heal.
**********************************************************************************************************
****I’m a dietitian, but probably not your dietitian. Please consult your healthcare provider before making changes to your diet, supplements, medications, or lifestyle. This is written for informational purposes and is not meant to diagnose or treat a condition. – Meredith Arthur, MS, RD, LD*****