
by Meredith Arthur, MS, RD, LD. This is not medical advice and is written only for informational purposes. Please consult with your provider before making changes to your diet, supplements, medication, or lifestyle.
********************************************************************************************************
As Jenny Jones, PhD recovered from acquired sulfite oxidase deficiency due to the MoCo Steal and overall decreased SUOX production due to inadequate vitamin B6 (pyridoxal phosphate), which is needed to make heme, a component of the sulfite oxidase enzyme, she noticed that when she increased her molybdenum intake, she experienced an oxalate dump. In contrast, my daughter Zoey is no longer excreting oxalate in appreciable amounts and has been deemed “cured” by her urology team because she no longer has oxalate-induced inflammation of ureters, and her kidney reflux has resolved, despite having ongoing health issues that would cause increased oxalate production in the body. Who is healthier? Jenny or Zoey?
For audio/visual learns, you can watch this video.
What is oxalic acid (oxalate)?
Oxalic acid is a small, naturally occurring dicarboxylic acid found in plants, fungi, and some bacteria. Plant foods have varying amounts. Some foods are high, and some foods are low. Marek Doyle has an online oxalate calculator if you would like any easy way to estimate how much dietary oxalate you consume per day. OXALATE CALCULATOR – MarekDoyle.com – Metabolic & Nervous System Co-Pilot. Cronometer can also estimate the amount of Oxalate you consume, although be sure to monitor the data confidence score, as not all foods list their oxalate content. The Trying Low Oxalate group, founded by Susan Owens, on Facebook can point you to a large Excel spreadsheet file that contains oxalate data. Other practitioners and groups that share extensively about the impacts of dietary oxalate include Sally Norton, Emily Givler, and Elliot Overton. You can refer to these practitioners for more details on dietary oxalate.
However, we also make oxalate in our liver cells from glycolaldehyde and hydroxyproline degradation, and I believe that sulfite toxicity severely alters oxalate metabolism and excretion!
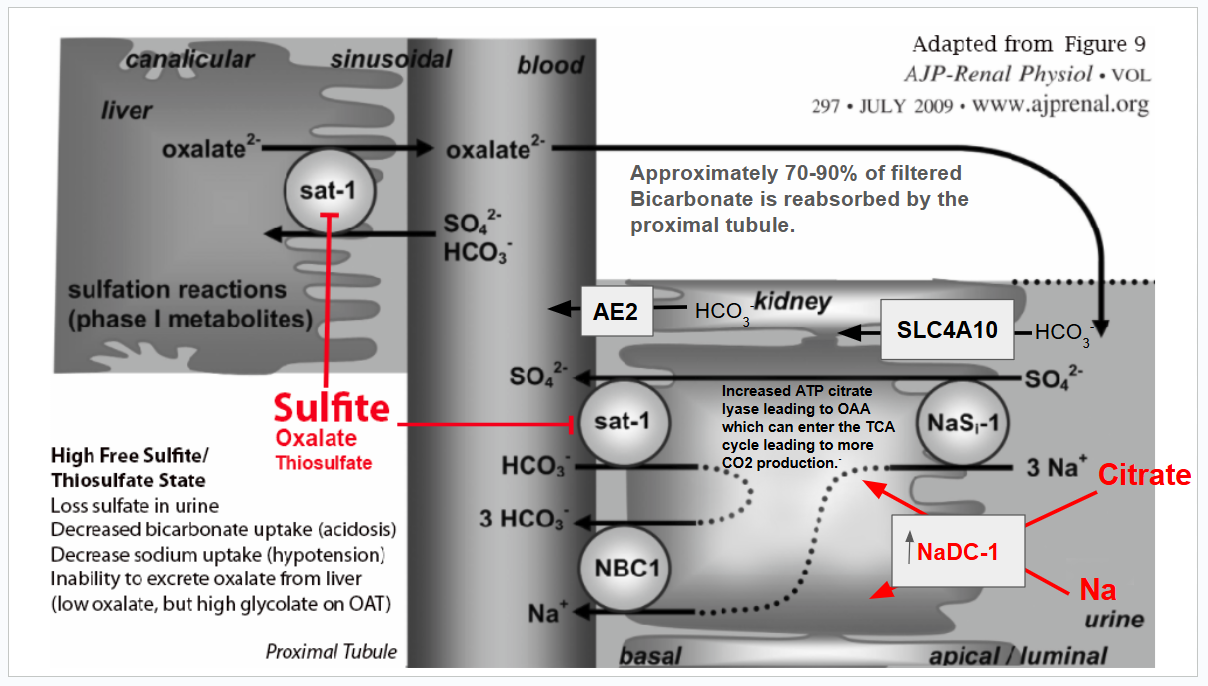
The following diagram summarizes the alterations that occur to oxalate metabolism and clearance during sulfite toxicity. Please read beyond this diagram for more information, references, and Zoey’s medical story, which prompted me to explore how sulfite and oxalate are tightly bound together.

Cofactors in blue are compromised by sulfite or cysteine deficiency. Prolonged sulfite exposure decreases NADPH by inhibiting glucose-6-phosphate dehydrogenase. (G6PHD) A functional B6 deficiency leads to a decrease in the de novo synthesis of NAD. Sulfite destroys B1. Sulfite can reduce lipoic acid levels by decreasing the total available cysteine when it binds to cystine, forming s-sulfocysteine (SSC). Sulfite forms an adduct with riboflavin, decreasing its catalytic activity. Sulfite can reduce CoA synthesis from pantothenic acid, as this requires cysteine that is tied up in SSC.
Jenny wins the “I have successfully recovered from sulfite toxicity award” due to oxalate dumping!
Jenny is by far healthier than Zoey, despite Zoey being discharged from urology (but I am hoping that nephrology will address what I think is going on based on her 24-hour urine test). Having previously explored the interactions between sulfite and oxalate metabolism, I was so excited for Jenny to report that she was experiencing oxalate dumping because an oxalate dump, to me, is a true sign of normalized sulfite oxidase function (usually). As I explain in the diagram above, oxalate dumps can occur when sulfite is mopped up, sulfate levels are increased, or when bicarbonate levels are increased. Jenny’s oxalate dump was due to restoration of sulfate and a decrease in sulfite levels, which restores oxalate production by releasing the inhibition of sulfite on glycolate oxidase, as well as allows more oxalate to be cleared because sulfite and thiosulfate are no longer inhibiting the movement of oxalate out of tissues via anion exchangers.
Thiamine supplementation can cause an oxalate dump!
However, not all oxalate dumps are related to healing! When I was still severely sulfite toxic, I had an oxalate dumping experience from adding 25 mg of thiamine HCl to 2 liters of water in an attempt to restore B1, as I suspected that sulfite had destroyed my B1. I had been struggling with the accumulation of fluid in my feet and ankles, which is a classic sign of heart failure. Thiamine deficiency is the only cause of heart failure, but ALDH2 deficiency is implicated in heart failure as well. Besides sulfite causing a thiamine deficiency by destroying thiamine, Sulfite exposure, whether from preservatives or endogenous production, generates sulfite radicals, which worsen when SUOX activity is decreased. Sulfite radicals cause oxidative stress that leads to post-translational inhibition of aldehyde dehydrogenase 2, ALDH2. ALDH2 deficiency can cause heart failure.
Suspecting that sulfite was causing a thiamine deficiency, I decided to start slowly and increase B1 intake, along with potassium citrate and magnesium citrate as cofactors for B1. I thought it plausible that I had wet Beriberi, a type of heart failure caused by thiamine deficiency. Below is a picture of my urine after one day of 25 mg of thiamine HCl while suffering from symptoms of sulfite toxicity. After this cloudy urine, I went to the emergency room with convulsions and internal tremors so severe that my teeth were chattering.

The internal tremors were most likely due to severely low ionized calcium. Unfortunately, ionized calcium levels were not checked at the emergency room. Instead, when I got to the emergency room, I was immediately checked for drug abuse as the tremors that I had started in my spine and moved outward. I looked as if I was having a drug overdose or withdrawal. I suspect that I was experiencing severe calcium dysregulation due to supplementation with potassium citrate and magnesium citrate (citrate binds to ionized calcium) as well as thiamine, inducing an “oxalate dump”. Thiamine can “mop up” sulfite, as when sulfite destroys thiamine, it actually forms an irreversible covalent bond.
When sulfite levels decrease, the inhibition of SAT-1 is released, and oxalate can move out of the liver in exchange for bicarbonate, chloride, or sulfate. During that emergency room visit, I was in hyperchloremic, non-anion gap acidosis, and chloride was likely serving as the anion exchanger for SAT-1 when sulfite levels decreased. Between the high amounts of oxalic acid in my blood, as well as supplemental citrate, ionized calcium levels likely plummeted. Luckily, I suspected I had hyperchloremia and asked that they not give me normal saline. Instead, I was given Lactated Ringer’s. Lactated Ringer’s contains 1.5 -3 mEq of calcium per liter. My tremors resolved! However, they did return on and off over the next several weeks and occasionally still occur due to the negative effects of elevated s-sulfocysteine on calcium regulation.
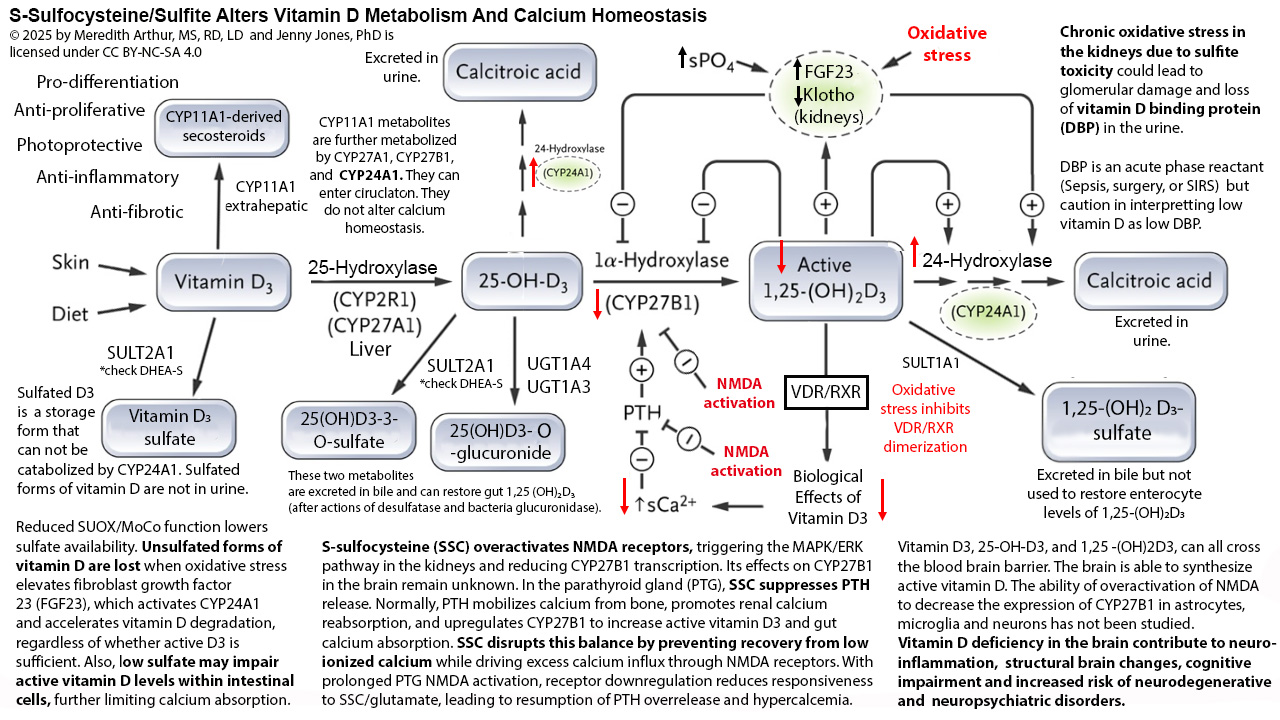
Below is a diagram that I have made to explain calcium dysregulation that occurs when sulfite accumulates and forms excessive s-sulfocysteine that acts like glutamate and can activate NMDA receptors in the kidney and parathyroid gland, causing alterations in calcium homeostasis. In the kidney, activation of NMDA receptors leads to decreased gene transcription for CYP27B1, resulting in decreased conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Typically, this decrease in active vitamin D eventually would lead to lower levels of calcium, but in response, the parathyroid gland would release parathyroid hormone (PTH) to trigger the release of calcium from bones, reuptake of calcium from the urine, and increase the activity of CYP27B1 in the kidneys. However, when NMDA receptors in the parathyroid gland are activated, PTH is not released. This leads to a state of hypoparathyroidism and low calcium.
No one should be excited about an oxalate dump!
Even though I was excited that Jenny was on the right road to recovery, she was not so excited about oxalate dumping, and rightly so. When oxalate begins to move out of tissues, it comes with disturbing symptoms such as crystals coming out of the tear ducts, stick-like structures coming out of pores, urinary tract irritation and burning, gastrointestinal distress, rashes, itching, and cloudy urine. Plus, it is possible that if kidney clearance of oxalate is poor, it can redistribute back into tissues in the body.
Many practitioners exist who you can turn to learn more about how to handle the symptoms of oxalate clearance. The purpose of this blog is to share the diagram below that explains how decreased SUOX function can lead to alterations in oxalate metabolism and to share my daughter’s ongoing dysfunction due to sulfite toxicity.
Zoey’s Oxalate Story
My daughter, Zoey, has struggled with high urinary oxalate levels on and off for most of her childhood, but since 2023, she has suddenly been “cured” of her hyperoxaluria per urology, except for the time she went to the emergency room with an adrenal crisis due to profuse vomiting after having two plantain muffins per day for four days. Hindsight revealed that this poisoning was due to a poorly written handout from a well-meaning dietitian who labeled plantains as low oxalate. Zoey has been on a low oxalate diet since age two after a Mosaic organic acid test showed urinary oxalate levels of 477 mmol/mol creatinine (normal range 6.8 -101 mmol/mol creatinine). I gave her plantain flour because her potassium was dropping very low, and plantain is a good source of potassium. However, instead of having 1 mg of oxalate per plantain, it can have up to 525 mg of oxalate per plantain. After she was released from the emergency room with “new onset cyclic vomiting syndrome” and a referral back to her current gastroenterologist, her labs were finally released to the online charting system. I was shocked to see that she had massive crystals in her urine and bladder. We were referred to metabolic genetics to check to see if she had primary hyperoxaluria, but they reported no gene alterations that would be pathogenic and result in high oxalate levels. Later, urology helped out by doing a 24-hour urinary stone risk assessment because my husband has a history of kidney stones.
But wait? What?! Zoey’s 24-hour urinary oxalate is low?!
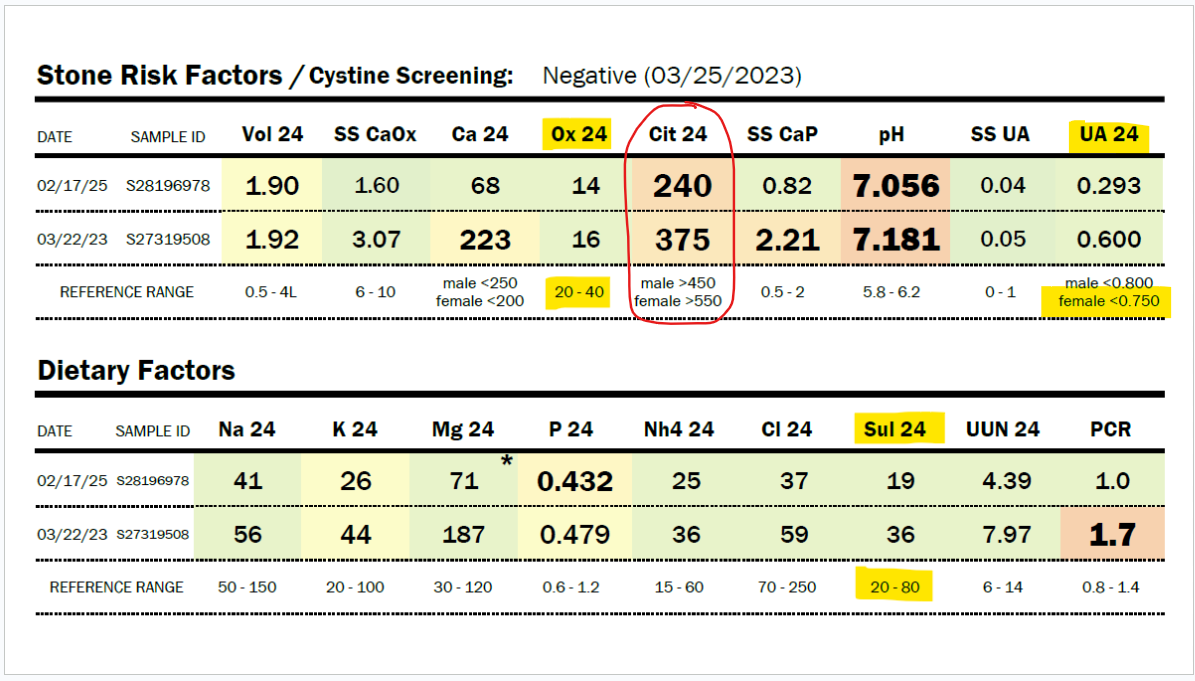
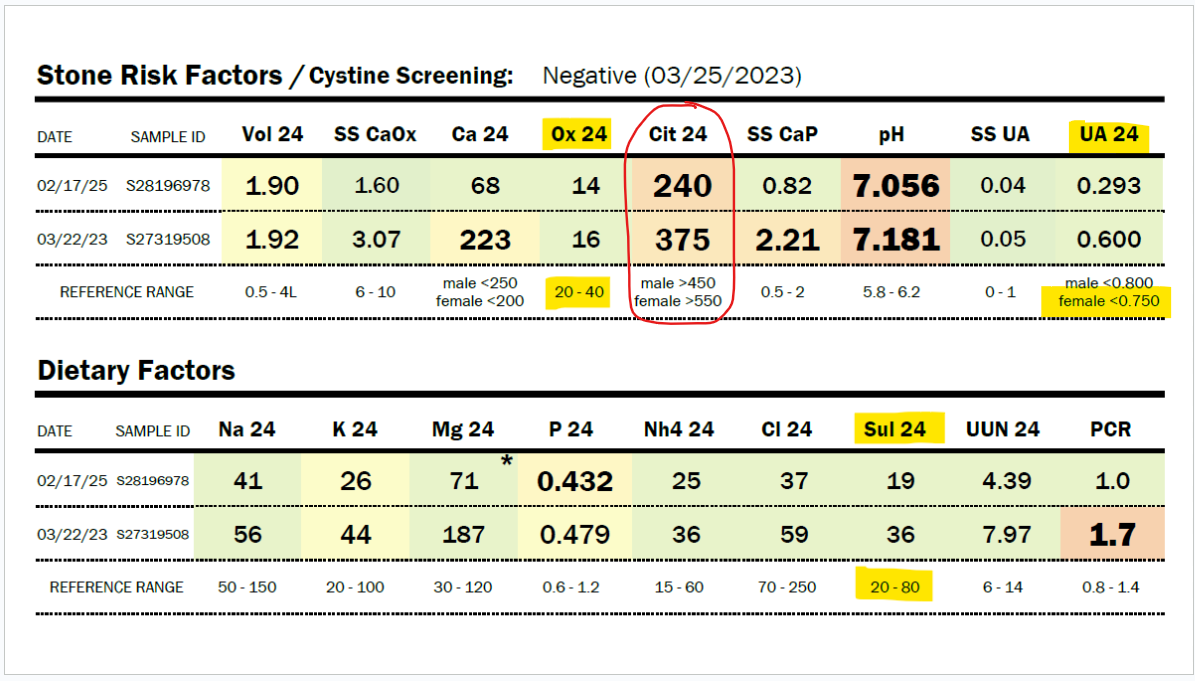
Due to her history of severe hyperoxaluria at the age of two years old, and a history of hydronephrosis, her physicians agreed to do a 24-hour urine collection, and she was found to have only 16 mg/24 hour of oxalate in 2023 (normal 20-40 mg/24 hour). In 2025, Zoey was found to have only 14 mg of oxalate in a 24-hour urine collection. Some might say that this just means she isn’t clearing oxalate in that 24-hour period, but Zoey’s stone risk assessment shows that the pH of her urine and her very low citrate levels would make her at risk for kidney stones. She has no stones. No evidence of oxalate leaving her body in excess.
My daughter has a history of sleep apnea. She should, in fact, be producing a large amount of oxalate daily, as sleep apnea increases myeloperoxidase activity, which increases the conversion of L-serine to glycolaldehyde, which becomes glycolate. Zoey has moderate to severe obstructive and central sleep apnea. Glycolate, as you can see in the diagram above, is converted to oxalate by the enzyme glycolate oxidase. In addition, sleep apnea increases the activity of xanthine oxidase. This should lead to very high uric acid levels, but Zoey’s 24-hour uric acid levels are below normal, as shown in the results immediately below this paragraph. They have decreased by half between 2023 and 2025, and she has not had improved sleep apnea. Also, her organic acid test in 2024 showed very high glycolate levels, but oxalate levels within the normal range on the Mosaic spot morning urine test (see below).
What’s going on with Zoey?
What I specifically think is happening to Zoey is that free sulfite toxicity in the liver can slow down oxalate production by inhibiting glycolate oxidase. Thiosulfate, which increases when sulfite oxidase isn’t working well, and sulfite can both inhibit the release of oxalate produced from hydroxyproline degradation. This causes the accumulation of oxalate in the liver. Having a below normal level of urinary oxalate on a 24-hour urine test or on an organic acid test is not necessarily a good thing. Having high glycolate and low oxalate certainly points to probable sulfite toxicity, as sulfite can decrease NADPH by inhibiting glucose-6-phosphate dehydrogenase, which is needed for GR/HPR conversion of glycolate to glyoxylate (which is metabolized to oxalate), as well as sulfite oxidase.
Basically, when sulfite is high, it means that whenever the body is increasing the production of glycolate, it is not being converted to glyoxylate, and also, when oxalate is produced from hydroxyproline degradation, the oxalate potentially stays in the liver. Zoey’s very low citrate levels could point to the fact that her liver is struggling with maintaining adequate movement of the TCA cycle. Oxalic acid can inhibit pyruvate carboxylase, leading to low levels of oxaloacetate and an overall decrease in citrate levels (acetyl CoA and oxaloacetate via the enzyme citrate synthase combine to form citrate).
However, in pyruvate carboxylase deficiency, typically, lactate and pyruvate are very high on organic acid testing. Hers was not, although this mosaic organic acid test was not done at the same time as the 24-hour urine test, so it could be possible that at the time of the 24-hour urine test, she was struggling more with pyruvate carboxylase deficiency. She also had normal lactate, pyruvate, and a normal L:P ratio.
Sulfite toxicity causes severe metabolic acidosis in the liver during sulfite oxidase deficiency. It could be at the time of this 24-hour urine test in March of this year, 2025, that she was suffering more from sulfite toxicity than when the Mosaic test was done in June 2024. You can see below her total urinary oxalate levels remain low (either from SAT-1 inhibition of oxalate release from the liver and/or sulfite inhibition of glycolate oxidase). She also has low uric acid levels, which is deemed normal, but due to hypoxia, Zoey should have high uric acid production from increased XO activity. And the number one issue that makes me believe that she has very low SUOX activity is the low urinary sulfate levels.
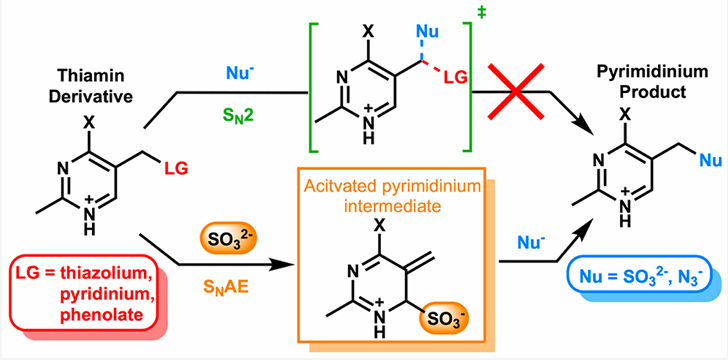
Although oxalate inhibition of pyruvate carboxylase is possible at the time of this 24-hour urine test, I suspect that the low urinary citrate is a byproduct of metabolic acidosis, coupled with SAT-1 inhibition by sulfite/thiosulfate. In response to any metabolic acidosis, whether SAT-1 is inhibited or not, the kidneys will increase NaDC-1, a sodium and citrate transporter that uptakes citrate from the urine while also increasing the expression of ATP citrate lyase. This enzyme converts citrate from the urine into OAA and acetyl-CoA in the cytosol of the proximal renal tubule. These are taken up into the mitochondria and metabolized through the TCA cycle. Isocitrate dehydrogenase and alpha-ketoglutarate dehydrogenase produce CO2, which helps to restore bicarbonate levels. Alpha-ketoglutarate dehydrogenase is a thiamine-dependent enzyme. Sulfite inactivates thiamine. (Sulfite-Catalyzed Nucleophilic Substitution Reactions with Thiamin and Analogous Pyrimidine Donors Proceed via an SNAE Mechanism | The Journal of Organic Chemistry) which could lead to less of a recovery from acidosis than should happen. In truth, Zoey is chronically in metabolic acidosis.
Zoey, The Suffering Sulfite Toxicity Trailblazer.
Why is she always struggling with sulfite?
Zoey has chronic obstructive and central sleep apnea, both of which contribute to the ongoing burden on sulfite oxidase and to the MoCo Steal that Jenny Jones, Ph.D. first described to me about two years ago. Obstructive sleep apnea increases xanthine oxidase activity, a MoCo-dependent enzyme. It also increases H2S production by the enzyme CBS and increases sulfite production from cysteine via CDO due to induction of the HIF-1alpha pathway. Zoey’s SUOX is highly burdened, and she appears not to have enough MoCo available at times, as evidenced by her lower uric acid levels.
My husband and I do our best to continuously put on her CPAP mask at night, but she takes it off throughout the night. We sometimes have a night nurse to assist with this, but not always. In addition, she has a genetic syndrome that truly alters her metabolism, including increasing the mRNA expression of MOCOS, an enzyme that converts MoCo to the form that is needed for AOX, XO, and XDH, but not the form needed by SUOX. (For my fellow MAND parents, you can find the alterations in mRNA expression of 448 genes in the supplemental files of this paper, Transcriptome analysis of MBD5-associated neurodevelopmental disorder (MAND) neural progenitor cells reveals dysregulation of autism-associated genes – PMC). MBD5-associated neuronal developmental disorder (MAND) is quite a puzzle to sort out. Although I may never get her fully out of sulfite toxicity, her suffering is helping others to find a way of escape.
Be thankful for Zoey.
Despite appearing normal from a urology standpoint, Zoey’s overall health is suffering from sulfite!
What concerns me the most and is now quite obvious to me after studying intensively about MoCo and SUOX after Jenny Jones, PhD, shared her MoCo Steal Theory of sulfite toxicity, is that Zoey does NOT have enough sulfate in her urine. Her urinary sulfate levels are below normal for her age, and in Autism we usually see urinary sulfate wasting, not low urinary sulfate levels. ((PDF) Sulphur Metabolism in Autism) I believe that Zoey is struggling with chronic sulfite toxicity and molybdenum cofactor deficiency to the point that she doesn’t even have enough sulfate available to experience sulfate wasting.
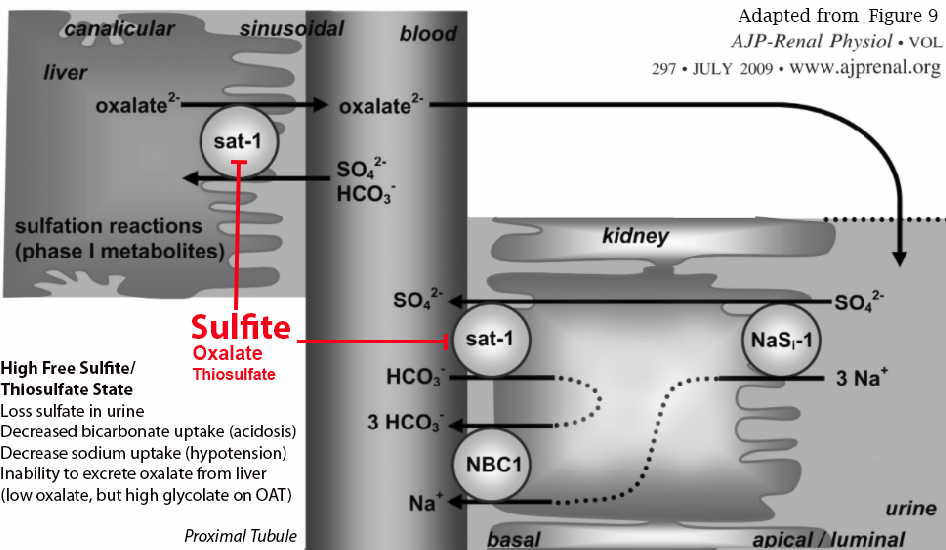
Diagram reference: Ability of sat-1 to transport sulfate, bicarbonate, or oxalate under physiological conditions
I have asked multiple doctors on her team to order a sulfite oxidase deficiency panel from Mayo Clinic. Still, I have received zero responses because it is not their area of expertise. They feel that if the results show sulfite oxidase deficiency, they won’t be able to provide a treatment plan. I do hope that our nephrologist will help by ordering the test from Mayo when we follow up in a few weeks. However, I’m confident that I am doing the best that I can to help Zoey with this issue.
As the 24-hour urine testing was last done in March of this year, I wanted to see if we have made any progress towards meeting Zoey’s nutrition needs to maximize the synthesis of molybdenum cofactor and sulfite oxidase. I recently sent her urine off to Diagnostic Laboratory Solutions to see if her SSC levels are high. On nights that Zoey has insomnia, which can be caused by SSC due to its acting like a glutamate in the brain, Zoey’s morning urine is at least 10 ppm on a water sulfite testing strip. However, I would like to see her SSC levels. I truly wish I had known about the OMX test from DLS sooner. I do hope that her SSC is not high now that we have worked on many cofactors to help with increasing the production of MoCo and SUOX. I will post an update when I get those labs back.
So, has Zoey “dumped” oxalate yet?
No, she has not.
Will I celebrate the day she does have an oxalate dump?
Yes, I will. This will mean that she is no longer sulfite toxic.
************************************************************************************
ALDH enzymes are inhibited by oxidative sSulfite and Thiosulfate Alter the Metabolic Processing of Oxalate © 2025 by Meredith Arthur, MS, RD, LD and Jenny Jones, Ph.D. is licensed under CC BY-NC-ND 4.0![]()
![]()
![]()
![]() tress-induced increases in GSSH, CysSSH, and exogenous persulfides such as garlic-derived trisulfides.
tress-induced increases in GSSH, CysSSH, and exogenous persulfides such as garlic-derived trisulfides.
- Is Aldehyde Dehydrogenase Inhibited by Sulfur Compounds? In Vitro and in Vivo Studies.
- A Regulatory Cysteine Residue Mediates Reversible Inactivation of NAD-Dependent Aldehyde Dehydrogenases to Promote Oxidative Stress Response.
- Redox Signaling Regulated by Cysteine Persulfide and Protein Polysulfidation.
- Reactive Cysteine Persulfides and S-Polythiolation Regulate Oxidative Stress and Redox Signaling.
- Mechanism of Sulfite Cytotoxicity in Isolated Rat Hepatocytes.
- Role of Aldehyde Dehydrogenases in Physiopathological Processes.
- Cross-Talk Between (Hydrogen)Sulfite and Metalloproteins: Impact on Human Health.
Alterations in SLC26 family of oxalate transporters by sulfite and thiosulfate
- Pathophysiology and Treatment of Enteric Hyperoxaluria.
- Oxalate Homeostasis.
- Ability of Sat-1 to Transport Sulfate, Bicarbonate, or Oxalate Under Physiological Conditions.
- Absence of the Sulfate Transporter SAT-1 Has No Impact on Oxalate Handling by Mouse Intestine and Does Not Cause Hyperoxaluria or Hyperoxalemia.
- Urolithiasis and Hepatotoxicity Are Linked to the Anion Transporter Sat1 in Mice
- Physiological Roles of Renal Anion Transporters NaS1 and Sat1.
- A Defect in Molybdenum Cofactor Binding Causes an Attenuated Form of Sulfite Oxidase Deficiency
- Isolated Sulfite Oxidase Deficiency
- Slc26 Family of Anion Transporters in the Gastrointestinal Tract: Expression, Function, Regulation, and Role in Disease
- The SLC26 Gene Family of Anion Transporters and Channels.
- The Enigmatic SLC26A6 Multifunctional Anion Transporter: Recent Advances in Structure-Function Relationship, Pathophysiological Significance and Novel Pharmacological Inhibitors
- Structural and Functional Properties of the Transporter SLC26A6 Reveal Mechanism of Coupled Anion Exchange
- SLC26 Anion Transporters.
- Sulfate and Thiosulfate Inhibit Oxalate Transport via a dPrestin (Slc26a6)-Dependent Mechanism in an Insect Model of Calcium Oxalate Nephrolithiasis.
- Characterization of Renal NaCl and Oxalate Transport in Slc26a6 Mice.
- Renal and Intestinal Transport Defects in Slc26a6-Null Mice.
- Essential Roles of CFEX-mediated Cl(-)-Oxalate Exchange in Proximal Tubule NaCl Transport and Prevention of Urolithiasis.
- Pathophysiology and Treatment of Enteric Hyperoxaluria.
- Regulation of the expression of the hepatocellular sulfate-oxalate exchanger SAT-1 (SLC26A1) by glyoxylate: a metabolic link between liver and kidney? – PubMed
Oxidative Stress Induces Metalloprotease, which Increases hydroxyproline catabolism
- Human Matrix Metalloprotease Activation by Insults of Bacterial Infection Involving Proteases and Free Radicals.
- Activation of Matrix Metalloproteinases by Peroxynitrite-Induced Protein S-Glutathiolation via Disulfide S-Oxide Formation.
- Mitochondrial Redox Control of Matrix Metalloproteinases.
- Oxidative Stress Augments the Production of Matrix Metalloproteinase-1, Cyclooxygenase-2, and Prostaglandin E2 Through Enhancement of NF-kappa B Activity in Lipopolysaccharide-Activated Human Primary Monocytes
- Redox-Sensitive Gene-Regulatory Events Controlling Aberrant Matrix Metalloproteinase-1 Expression.
- Hydrogen Peroxide (H2O2) Increases the Steady-State mRNA Levels of Collagenase/MMP-1 in Human Dermal Fibroblasts.
- Regulation of Matrix Metalloproteinases by Cytokines and Reactive Oxygen/Nitrogen Species in the Myocardium.
- Catalase Restores the Altered mRNA Expression of Collagen and Matrix Metalloproteinases by Dermal Fibroblasts Exposed to Reactive Oxygen Species.
- Molecular Mechanism for Activation and Regulation of Matrix Metalloproteinases During Bacterial Infections and Respiratory Inflammation.
- Collagenase Expression and Activity Is Modulated by the Interaction of Collagen Types, Hypoxia, and Nutrition in Human Lung Cells.
- Matrix Metalloproteinase Collagenolysis in Health and Disease.
- Proline Metabolism and Microenvironmental Stress.
NMDA receptor overactivation can lead to increased collagen degradation by inducing 1,25 OH-D deficiency
- Sustained Activation of Renal N-Methyl-D-Aspartate Receptors Decreases Vitamin D Synthesis: A Possible Role for Glutamate on the Onset of Secondary HPT.
- Vitamin D Deficiency.
- Vitamin D Physiology.
- Bone Resorption Markers in Vitamin D Insufficiency.
- Vitamin D for the Prevention of Disease: An Endocrine Society Clinical Practice Guideline.
- S-Sulfocysteine/NMDA Receptor-Dependent Signaling Underlies Neurodegeneration in Molybdenum Cofactor Deficiency.
- Molybdenum Cofactor Deficiency.
Hypoxia Inducible Factor One Alpha Related to Hydroxyproline and Oxalate Production
- HIF1α Is a Central Regulator of Collagen Hydroxylation and Secretion Under Hypoxia During Bone Development.
- Hypoxia-Inducible Factor Prolyl 4-Hydroxylases: Common and Specific Roles.
- Proline Hydroxylation and Gene Expression.
- Structural Basis for the Recognition of Hydroxyproline in HIF-1 Alpha by pVHL.
- Hypoxia-Inducible Factor-1 (HIF-1).
- Modulation of the Hypoxic Response.
- Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous?.
- Cellular Signal Transduction of the Hypoxia Response.
- The Regulatory Mechanisms of Proline and Hydroxyproline Metabolism: Recent Advances in Perspective.
- The New Insight Into the Role of Hydroxyproline in Metabolism of Cancer Cells.
- Proline Metabolism and Microenvironmental Stress.
- Global Metabolic Profiling Identifies a Pivotal Role of Proline and Hydroxyproline Metabolism in Supporting Hypoxic Response in Hepatocellular Carcinoma.
B1 acts as a sulfite mopper upper (and is destroyed by sulfite)
Low citrate could be caused by oxalate inhibition of pyruvate carboxylase
- The Clinical and Biochemical Implications of Pyruvate Carboxylase Deficiency.
- Pyruvate Carboxylase Deficiency: Mechanisms, Mimics and Anaplerosis.
- Inhibitors of Pyruvate Carboxylase – PMC
- Metabolic effects of oxalate in the perfused rat liver – PubMed
Sleep apnea increases XO, HIF-1alpha, CDO and CBS
- Hypoxia-inducible factors and obstructive sleep apnea – PMC
- Hypoxia Inducible Factors and Hypertension: Lessons from Sleep Apnea Syndrome – PMC
- HIF-1α Activation by Intermittent Hypoxia Requires NADPH Oxidase Stimulation by Xanthine Oxidase – PMC
- Long-Term Intermittent Hypoxia Elevates Cobalt Levels in the Brain and Injures White Matter in Adult Mice – PMC
- Xanthine oxidase mediates hypoxia-inducible factor-2α degradation by intermittent hypoxia – PubMed
- Hypoxia-inducible factors regulate human and rat cystathionine β-synthase gene expression – PubMed
- Hypoxia-inducible factor induces cysteine dioxygenase and promotes cysteine homeostasis in Caenorhabditis elegans | eLife
Sulfite inhibits G-6-Phosphate Dehydrogenase
- Bezafibrate Prevents Mitochondrial Dysfunction, Antioxidant System Disturbance, Glial Reactivity and Neuronal Damage Induced by Sulfite Administration in Striatum of Rats: Implications for a Possible Therapeutic Strategy for Sulfite Oxidase Deficiency.
- The Mitochondrial-Targeted Reactive Species Scavenger JP4-039 Prevents Sulfite-Induced Alterations in Antioxidant Defenses, Energy Transfer, and Cell Death Signaling in Striatum of Rats.
- In Vitro Evidence That Sulfite Impairs Glutamatergic Neurotransmission and Inhibits Glutathione Metabolism-Related Enzymes in Rat Cerebral Cortex.
ALDH2 deficiency and heart failure.
- Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling – PubMed ;
- Aldehyde dehydrogenase 2 and arrhythmogenesis
- Cardiac Mitochondrial Respiratory Dysfunction and Tissue Damage in Chronic Hyperglycemia Correlate With Reduced Aldehyde Dehydrogenase-2 Activity.
- Aldehyde Dehydrogenase 2 Activation in Heart Failure Restores Mitochondrial Function and Improves Ventricular Function and Remodelling.
- Mitochondrial Aldehyde Dehydrogenase and Cardiac Diseases.
The kidneys response to metabolic acidosis by increasing citrate uptake and metabolism.
- Acid-Base Transport by the Renal Proximal Tubule.
- Molecular Pathophysiology of Acid-Base Disorders.
- Structure, Function, and Regulation of the SLC4 NBCe1 Transporter and Its Role in Causing Proximal Renal Tubular Acidosis.
- Physiologic and Molecular Aspects of the Na+:HCO3- Cotransporter in Health and Disease Processes.
- Proximal Tubule Function and Response to Acidosis.
Glutamate/S-sulfocysteine alters in calcium homeostasis by altered CYP27B1 and PTH release
- Sustained Activation of Renal N-Methyl-D-Aspartate Receptors Decreases Vitamin D Synthesis: A Possible Role for Glutamate on the Onset of Secondary HPT.
- Hypercalcemia: A Review.
- N-Methyl-D-Aspartate Receptors Are Expressed in Rat Parathyroid Gland and Regulate PTH Secretion.
- Genomic Mechanisms Controlling Renal Vitamin D Metabolism.