Zoey’s metabolome showed increased urinary losses of biopterin. I was asked by the research scientist if I was supplementing with biopterin at the time of submitting urine samples. I was not. I suspect the cause of her losses of urinary biopterin are due to the damaging effects of sulfite toxicity on qBH2. Sulfite can bind to quinone dihydrobiopterin causing decreased recycling of qBH2 to BH4.
I’ve recently updated this diagram to include that choline oxidase is inhibited by sulfite. This is of major concern in sulfite toxicity due to betaine aldehyde can detoxify sulfite.
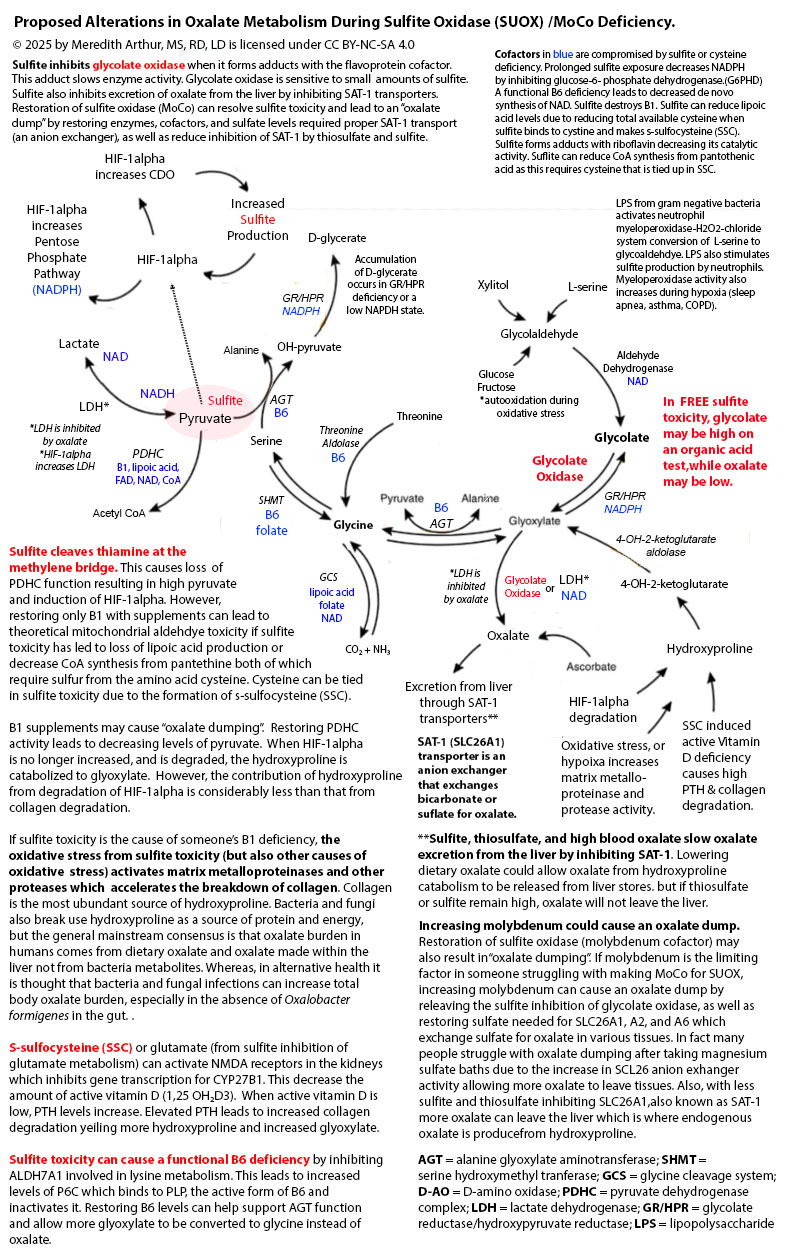
Zoey’s suddenly “cured” of hyperoxaluria as well as hyperuricemia. Is this a good thing? I think not. She has elevated glycolate levels, but low oxalate levels on her organic acid test, and her 24-hour urine oxalate levels are below the standard for someone her age as well as her urinary sulfate levels. I suspect worsening sulfite toxicity as the cause of her decreased glycolate to oxidase conversion. This isn’t a sign of improvement. It’s a sign of severe dysfunction. See the diagram below. Glycolate oxidase is inhibited by sulfite.
Zoey struggles with sleep apnea. Despite our best efforts, she rarely keeps her mask on for more than 2-3 hours per night. This means that during the night, her body increases gene transcription for HIF-1alpha in response to hypoxia. In a hypoxia state, HIF-1alpha goes to the nucleus and alters metabolism, including increasing the enzyme cysteine dioxygenase. This leads to a downstream increase in sulfite production which puts a burden on sulfite oxidase, the enzyme that metabolizes sulfite to sulfate. In addition, sleep apnea increases the activity of the enzyme xanthine oxidase. This used to be the cause of Zoey’s high uric acid levels. Xanthine oxidase uses molybdenum cofactor. Overall, sleep apnea sets the stage for Jenny Jones, PhD’s “Moco Steal” by increasing the need for more molybdenum for xanthine oxidase as well as increasing the total amount of sulfite produced by increasing CDO activity.
When the MoCo Steal from sleep apnea became extreme to the point that there wasn’t adequate molybdenum cofactor for sulfite oxidase, the sudden surge in sulfite caused a functional B6 deficiency through inhibition of ALDH7A1 resulting in a build-up of P6C. This led to decreased activity of cystathionine-beta synthase and cystathionine-gamma lyase. These two enzymes provide a significant amount of hydrogen sulfide that turns off the HIF-1alpha gene transcription. As hydrogen sulfide production decreased in Zoey’s enterocytes, she developed small intestinal bacteria overgrowth to compensate for the decrease in H2S (Greg Nigh has a theory that we grow sulfur metabolizing bacteria to provide our bodies what we need).
With the excess hydrogen sulfide in her gut, and a very oxalate restricted diet due to a history of hyperoxaluria, Zoey’s total molybdenum absorption decreased significantly due to sulfur can bind to molybdenum and when complexed with dietary copper, form a molybdenum-copper-sulfate complex that is unabsorbable. This perpetuated the problem. Over a year, her body became severely molybdenum cofactor deficient to the point that sulfite in her liver inhibited glycolate oxidase resulting in markedly high glycolate levels on her organic acids, but within range values of oxalate and below level oxalate on her 24-hour urine test.
Above you can see that Zoey’s 24-hour calcium levels have come down. She actually has decreased hypercalcemia because overall, she has improved from a vitamin A standpoint. Her serum vitamin A has dropped to 49 ug/dl which decreases osteoclasts being aggravated and breaking down bone.
Her 24 hour citrate levels have plummeted further. This is because sulfite can bind to pyruvate and prevent pyruvate metabolism to acetyl CoA. In addition, sulfite can damage all of the cofactors needed for the pyruvate dehydrogenase complex (B1, FAD, NAD, lipoic acid, and CoA). In addition, if sulfite is bound to pyruvate, it can’t be made into oxaloacetate. This leads to decreased overall levels of citrate.
Zoey’s urinary pH is quite neutral, but this is of concern to me because her blood chemistry panels over the past two years have shown hyperchloremic normal anion gap acidosis. This is consistent with renal tubular acidosis OR the actions of glutamate/s-sulfocysteine on NMDA in the kidney and activation of the sodium exchanger (ENaC). I have seen this hyperchloremic normal anion gap acidosis in many people who have suspected sulfite oxidase/moco deficiency. Activation of the ENaC results in uptake of sodium and chloride and wasting of potassium (sodium and chloride in urine below the reference range). Zoey’s urinary potassium is quite low, but her blood potassium is also low on blood draws. She has become potassium deficient and so her body is attempting to retain as much potassium as possible. During this 24-hour urine test she was getting 1000 mg of potassium citrate and 350 mg of magnesium citrate.
Zoey’s low phosphorous level is of a concern due to it may indicate intestinal malabsorption. She has a high risk for Crohn’s disease due to MBD5 deletion can increase the mRNA expression of FOLH1, that is higher in individuals with Crohn’s. The drop in her urinary sulfate below normal is consistent with sulfite oxidase deficiency. I suspect her levels are even lower because I give her magnesium sulfate foot baths every other day. The drop in her creatinine and urinary urea nitrogen indicated struggles with creatinine production (functional B6 deficiency and/or methylation decreased due to sulfite binding to B12, inhibiting betaine production, and oxidative stress damaging 5-methylfolate). Her low urinary urea nitrogen indicates a probable decrease in ornithine production needed for running the urea cycle as this is a byproduct of creatine production as well as can be made from proline but requires vitamin B6.
As you can see above, overall, Zoey is struggling with sulfite toxicity. I think that since birth (she had hypoxia in the womb due to placental abruption) Zoey has been stuck in the HIF-1alpha pathway to varying degrees her whole life. We are working on a therapeutic plan to deal with this constant, chronic uptick in sulfite production as well as strategic avoidance of sulfur foods at the time of taking mozyme forte.
References:
Ghanem, Mahmoud. On the mechanistic roles of the protein positive charge close to the N(1)flavin locus in choline oxidase.
Meier, Sebastian & Solodovnikova, Natalia & Jensen, Pernille & Wendland, Jürgen. (2012). Sulfite Action in Glycolytic Inhibition: In Vivo Real-Time Observation by Hyperpolarized 13C NMR Spectroscopy. Chembiochem : a European journal of chemical biology. 13. 2265-9. 10.1002/cbic.201200450.
But what is vitamin A toxicity? Do we all have full livers? No. 30% of us might have full livers.
Some of us have vitamin A dysregulation which is caused by poor metabolism (low NAD state) and/or effluxing vitamin A back into the blood due to high intracellular calcium (SSC activation of NMDA, EMF activation of calcium channels, Magnesium deficiency causes STRA6 to prefer to stay on and efflux vA out into blood). High blood vitamin A levels aren’t good for sure, but they are also a squeaking wheel for major metabolic issues.
If you have HIGH retinol and LOW uric acid you have a molybdenum cofactor deficiency, and this could merely be inadequate molybdenum intake. If you take molybdenum, but still have low uric acid and high retinol, then you are struggling with making MoCo. Come join the Moco Steal group. We need to be making MoCo to have back up enzymes for metabolizing retinol and retinaldehyde. It requires several cofactors to be made.
Does vitamin A, in the form of retinol, trigger us to make uric acid? No. I would say that retinol is associated with high uric acid first due to enzyme competition. If XOR family enzymes are busy dealing with other substrates on the left of the diagram below (caffeine, high dose niacin, purines, aldehydes, etc), then it can’t play back up for ALDH enzymes metabolizing retinol and retinaldehyde.
We need MoCo for SUOX which metabolizes sulfite. Sulfite destroys thiamine. Sulfite toxicity will cause the pentose phosphate shunt pathway to prefer to send RP5 towards uric acid. If sulfite toxicity is very high it will stop the pentose phosphate shunt pathway all together by inhibiting the first enzyme, glucose-6-phosphate dehydrogenase. Having low uric acid could mean severe sulfite toxicity and severe MoCo deficiency. Having high uric acid could mean low B1 status.
B1 Deficiency Causing Increase Purine Synthesis. Another possibility is that the underlying issue is excessive shuttling or RP5 towards purine synthesis in the setting of thiamine deficiency (needed for TKT activity) due to sulfite toxicity. This sulfite toxicity could have been originally caused by high dose vitamin A causing a MoCo Steal. Sulfite immediately destroys B1, but the body will do it’s best to mop up sulfite (binds to B12, betaine aldehyde, cysteine). However, B1 deficiency can be caused by other factors too including excessive caffeine intake, too much sushi, Lasix therapy, poor oral intake, etc. B1 deficiency causes more RP5 resulting in more PRPP and the production of more uric acid. This pulls XOR enzymes away from retinol and retinaldehyde metabolism.
Does a ZERO vitamin A diet solve the uric acid problem? No. In fact, it could make it worse. The adaptative immune system uses vitamin A in the form of retinoic acid (see diagram). We do need physiological levels of retinoic acid to prevent XOR dominance happening where the innate immune system attacks bacteria and viruses with H2O2 made using Xanthine Oxidase instead of recruiting additional immune cells.
Consuming 25-50% of the RDA of vitamin A plus adding a lower dose of niacin (35 mg) at those meals seems to help my family.
****************************************
I’m a dietitian, not a doctor. I am probably not your dietitian. Please consult with a personal healthcare provider prior to making changes to you diet, medications, supplements, or lifestyle. This is written only to inform, and not to diagnose or treat a condition. This blog does not replace an evaluation by a medical professional. – Meredith Arthur, MS, RD, LD
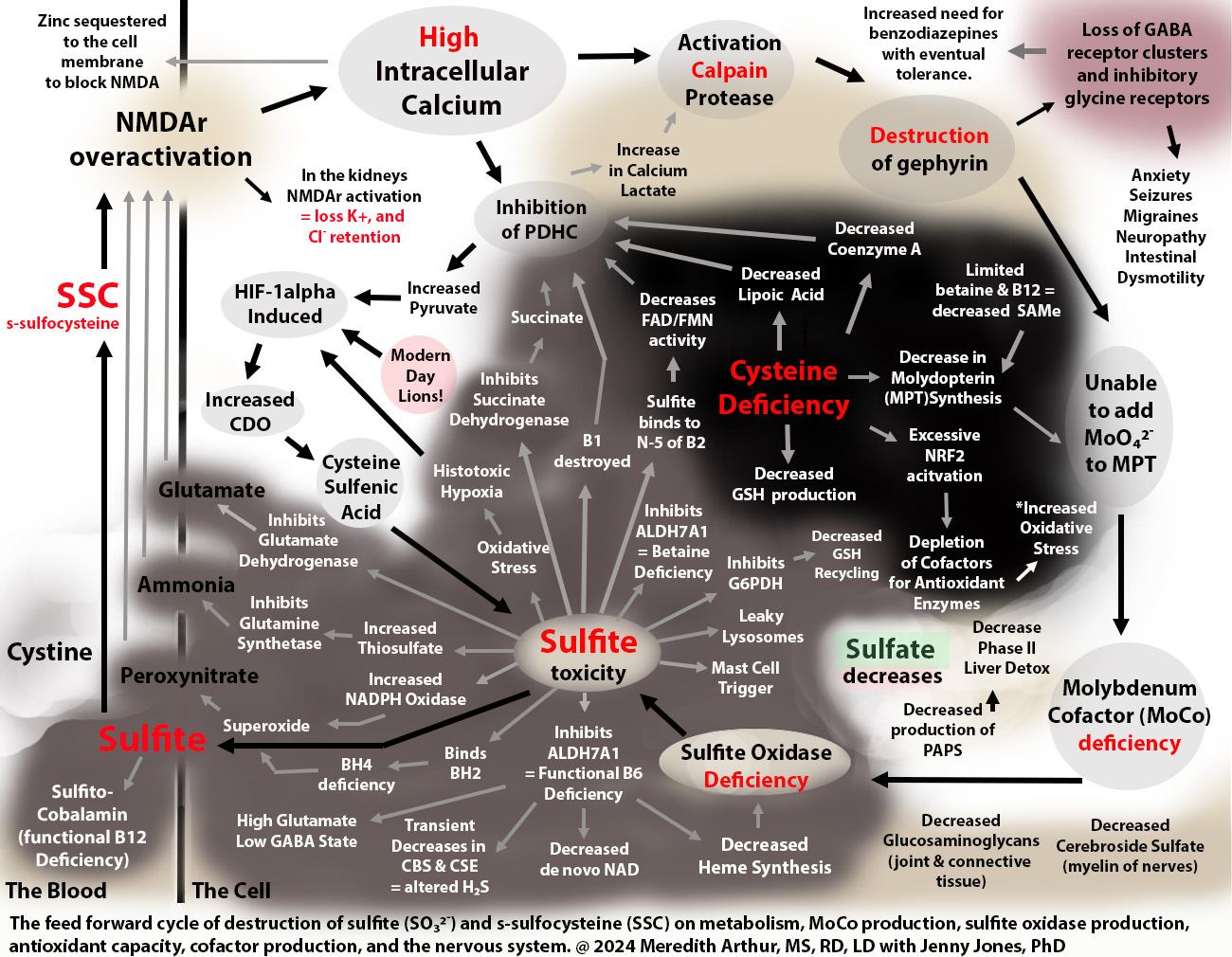
Jenny Jones PhD and I have a hypothesis paper explaining how a MoCo Steal can lead to a Sulfite Trap involving the excess production of s-sulfocysteine (SSC), an NMDA agonist on steroids, and excess sulfite production. The MoCo Steal was a term adopted by Jenny as she liked your phrase, “NADPH steal” and felt her cascade into seizure disorder was caused by a shift of molybdenum cofactor away from SUOX towards AOX due to inadequate NAD for working ALDH enzymes as well as other blocks to this including high dose vitamin C. My daughter Zoey has MBD5 deletion which leads to increased expression of Mocosulfurase which may have caused her preferences to make more xanthine oxidase and aldehyde oxidase and less sulfite oxidase. As we continued to research, we found that the sulfite toxicity can be caused by the interaction of the immune system and hypoxia pathway, hypoxia-inducible factor 1 alpha which increases the expression of CDO to help with s-sulfenylation during oxidative stress as a mechanism by which to protect cells, but that instead of this metabolic switch ending in sulfate, individuals struggling with the MoCo Steal, end up in a Sulfite Trap.
Once someone is not working SUOX properly, there is a feed-forward mechanism because SSC’s robust effect on NMDA causes intracellular calcium overload leading to activation of calpain protease with subsequent degradation of gephyrin. Gephyrin is needed to add molybdenum to the molybdenum cofactor (MoCo).
Damaging effects of SSC due to excess calcium influx
loss of gephyrin and ability to incorporate molybdenum into MPT
loss of gephyrin that holds GABA receptors in place resulting in loss of functioning GABA synapses and subsequent imbalance of glutamate to GABA with effects downstream neurological effects
inhibition of PDHC and lactic acidosis
inhibition of glutamate aspartate transporter (GLAST) causing loss of glutamate uptake in neurons resulting in low brain energy levels
potassium and sodium wasting via over-excitation of NMDA in the kidneys with subsequent loss of the ability to uptake bicarbonate, increased H+ ions, and retention of chloride causing a non-anion gap hyperchloremic acidosis
Sulfites direct damaging effects include:
need for metabolizing by cytochrome C using oxygen leading to loss of mitochondrial oxygen and production of sulfite radicals. The oxidative stress triggers HIF-1alpha which increases CDO and leads to more sulfite due to the loss of SUOX activity
inhibition of ALDH7A1 with subsequent altered lysine metabolism resulting in a build-up of P6C and functional B6 deficiency
cleavage of the methylene bridge of thiamine causing thiamine deficiency
binding to B12 as sulfito-cobalamin causing a functional B12 deficiency and loss of MUT causing inadequate succinyl CoA and slowed heme synthesis
inhibiting glutamate dehydrogenase in neurons results in decreased alpha-ketoglutarate, diminished movement of the TCA cycle, decreased mitochondrial membrane potential, and decreased ATP synthesis.
inhibiting glutamate dehydrogenase in other cells leads to similar effects, but also loss of alpha-ketoglutarate to act as an antioxidant during oxidative stress
damage to lysosomes causing them to leak cyanide which ties up MPST in the production of thiocyanate. Thiocyanate causes loss of uptake of iodine into the thyroid gland and hypothyroidism
Functional B6 deficiency from ALDH7A1 inhibition causes:
inability to complete de novo NAD production via the kynurenine pathway
loss of CBS and CSE activity
inability to start heme synthesis due to ALAS requires B6 (or minimal ability due to triage flux of B6 towards important cellular needs)
decreased serine hydroxy methyltransferase (SHMT) activity leading to decreased glycine production from serine, but glycine supplements cause reactions due to acting as co-agonists to NMDA
severe cases lead to a lack of histamine production (resolution of histamine intolerance but at the same time severe neurological presentation with loss of motor coordination)
less severe cases, decreased ability to metabolize histamine with ALDH7A1 side of histamine metabolism leading to increased burden on HNMT
decreased glutamate to GABA conversion
Sulfite binds to cystine to make SSC which can cause cystine deficiency. This could be restored by SSC being taken up by cells and metabolized using glutathione, but the shift towards the hypoxia pathway causes more cysteine to be shuttled towards CDO and overall there is a glutathione deficiency. Due to oxidative stress, cysteine is also shuttled towards CDO to produce cysteine sulfenic acid to protect enzymes during oxidative stress so that when it resolves the cell can resume function.
Cysteine deficiency then causes…
loss of lipoic acid, CoA, and FAD and FMN (sulfite binds to the N-5 catalytic site of B2 causing flavin containing enzymes to be nonfunctional).
overactivation of NRF2 activation leading to keratinization
vitiligo due to failure to prevent apoptosis of melanocytes and due to sulfite can inhibit glycolate oxidase. Glycolate inhibits tyrosine hydroxylase needed to produce melanin.
(CoQ10 needs cysteine for production – prenylation- but this seems conserved due to needing large amounts of ubiquinol for degradation of HIF-1apha)
Sulfite toxicity and cysteine deficiency collide at the crossroads of transsulfuration and the HIF-1alpha pathway.
I’m a dietitian, not a doctor. This isn’t medical advice. Talk with your healthcare provider before making changes to your diet, supplements, medication, or lifestyle.
For the past year or so Jenny Jones and I have been discussing her MoCo Steal Theory. Her theory is that due to blocks in NAD recycling and various other blocks at ALDH, she had to use more of the low affinity, high-capacity enzyme for vitamin A metabolism, aldehyde oxidase (AOX). This caused a stealing of molybdenum cofactor towards AOX and away from sulfite oxidase (SO or SUOX) leading to a loss of sulfite oxidase activity. Sulfite then caused the functional B6 deficiency that she and I collaborated on where the moonlighting enzyme ALDH7A1 was completely inhibited by sulfite causing a sort of late-onset pyridoxine-dependent epilepsy from a functional B6 deficiency.
.
Controlling glutamate is important through dietary interventions to reduce free glutamate if you struggle with seizures or migraines, but there is a sneaky glutamate-like compound being made in our bodies every day called s-sulfocysteine (SSC). It’s the dipeptide cystine bound to free sulfite. Chris Masterjohn talks of how we all have this in varying amounts.
.
Jenny, Andrew Baird, Michelle Harris, and I have been discussing that vitamin A “toxicity” is actually vitamin A “dysregulation” and overall isn’t a poisoning from retinoic acid because most of us aren’t even getting to the point of making retinoic acid due to the massive dysfunction caused by sulfite toxicity.
.
After many months of research, I have concluded that Jenny’s MoCo Steal may have happened to some of my clients in the past including my daughter. However, I noticed that they had gone beyond a MoCo Steal and had fallen into a Sulfite Trap because when we can’t metabolize sulfite, we make s-sulfocysteine (SSC) and have excess free sulfite causing damage to our mitochondria leading to a feedforward cycle of destruction that meets at the crossroads of transsulfuration and the immune and hypoxia response pathway, hypoxia-inducible factor-1 alpha (HIF-1alpha).
.
Regarding vitamin A metabolism. SSC over-activates NMDA receptors on many cells in the bodycausing large increases in intracellular calcium which then causes retinol to efflux out of cells back onto RPB4. This is the high serum A that we experience. It’s a cellular deficiency of vitamin A, but blood toxicity.
.
Although high liver vitamin A stores are still possible, I think that high serum vitamin A is the squeaking wheel on a very broken race car called Acquired Sulfite Oxidase Deficiency. This leads to intolerance to vitamin A-rich foods. The thesis linked to in the description section of the video will explain dry skin, collagen loss, and poor glucuronidation, but this is not covered in this preliminary video below which only explains how we get trapped in this pathway.
But the damage doesn’t stop at vitamin A intolerance.
Both sulfite in the mitochondria and SSC cause an increase in the activity of cysteine dioxygenase (CDO) through the activation of HIF-1alpha. Upregulation of CDO leads to shutdown of the cells through s-sulfenylation with cysteine sulfenic acid. This is a life-saving effort to protect enzymes from oxidative stress so that when the cell turns back on, the cell won’t have to start all over again. The bad part is, that leftover cysteine sulfenic acid is metabolized to sulfite and should be further metabolized to the beautiful, life-saving sulfate which is used for Phase II liver detox. Instead, we make more sulfite which leads to more mitochondrial damage as well as more SSC which causes more increases in CDO and more sulfite. It’s a trap.
.
SSC itself causes alterations in metabolism that cause the loss of the ability to make MoCo because it leads to loss of gephyrin needed to add molybdenum to molybdopterin and eventually loss of the ability to make the SUOX enzyme as well for some people. Cells outside of the central nervous system express NMDA receptors, which can respond to SSC, including hepatocytes, a major site of sulfite oxidase activity.
.
This pathway leads to cysteine deficiency from tying up cystine in the blood with sulfite. This eventually leads to the inability to make FAD due to FAD synthase needs the cofactor molybdopterin (MPT) which requires a sulfur group from cysteine. MPT is NOT molybdenum. It’s the step before molybdenum. Taking molybdenum is not a way to restore FAD synthesis. It’s not involved in that reaction. Just MPT, the empty shell of MoCo, for lack of better terms, is what is needed for making FAD.
.
And the damage to other cofactors and pathways is evident. Anything that requires cysteine will be decreased such as lipoic acid, coenzyme A, activation of NRF2, and production of CoQ10 (although I mostly see this pathway conserved, if not high on Genova testing, because people need CoQ10 for the HIF-1alpha pathway, but they aren’t using it in their mitochondria at SQR as the mitochondria are shut down trying to repair the damage from sulfite and I doubt that CBS and CSE are working much at all due to functional B6 deficiency from sulfite inhibiting ALDH7A1).
.
Activation of NMDA receptors in the kidneys causes potassium wasting and sodium wasting, but chloride retention, and inability to uptake bicarbonate from urine lead to non-anion gap acidosis which is often missed by doctors. It can also cause hyperaldosteronism and hypertension. I suspect that anyone requiring high dose thiamine for life with high dose potassium intake is a maker of excessive amounts of SSC. Free sulfite is probably the cause of many individuals’ thiamine deficiencies as sulfite cleaves thiamine at the methylene bridge essentially destroying it.
.
In the brain, SSC over-activates NMDA receptors on GABAergic synapses leading to excessive calcium influx into cells, loss of gephyrin needed to hold GABA receptors in place, and destruction of the synapses. This leads to a high excitatory state with low inhibition and can cause seizures as well as anxiety, pain, bowel dysmotility, tremors, and dysautonomia.
.So…I humbly ask for you to consider this hypothesis that I’m living out in real time. I am not trying to be a savior. I’m just sharing an awful experience and showing my love for my fellow humanity by putting this out there. I am currently stuck in the sulfite trap with my daughter, Zoey. I got here when I took garlic and NAC to lower my blood pressure and then added in rolled oats. It was a snowball effect leading to severe sulfite toxicity. I think I have a way of escaping, but I am tired.
Because of the underlying sympathetic surges that I have right now, negative comments will harm me physically. Please refrain from making negative comments about Jenny and my work until you have thoroughly investigated it and proven it wrong. I would like for all the great minds out there in our world to take over and help find a solution to this problem if they feel that my hypothesis is valid. I think I have a few good ideas, but I do not know everything. I need YOU…smart people. Help save my brain and everyone else’s, please.
,
If I am wrong on this HYPOTHESIS. I may end up on 20 medications to control the massive symptoms that I have. However, currently, Benadryl is helping. This is not a medication recommendation for anyone…you MUST go to your doctor or practitioner with this idea. This is a dangerous trap.
Benadryl blocks NMDA receptors nicely and is reducing all my symptoms, but I’m still not fixed because when I stop the Benadryl, I fall back into horrible symptoms such as anxiety, tachycardia, insomnia, high blood pressure (some have low blood pressure), pain, internal tremors, burning sensation all over my body, and more. Only time will tell if blocking NMDA digs me out of the trap. I have a few other ideas as well in the collaborative thesis paper that Jenny and I worked on.
If you aren’t having a hypertensive crisis, there is another way out. I had a client heal with a few small changes. She still has migraines and histamine reactions, but her neurological symptoms have improved immensely. More on that soon.
CAUTION!!!!!!
Do not try to get out of this trap alone!!! Blocking NMDA restarts metabolism and some things might happen that aren’t pleasant. If you over block NMDA and then remove the block, you could have a SEIZURE. You must be aware of this risk. PLEASE WORK WITH YOUR PRACTITIONER.
Do not add NAC if you think you are stuck in this pathway until you get with a practitioner to find some way to block your NMDA receptor UNDER the SUPERVISION OF A HEALTHCARE PRACTITIONER but titrate it for your own needs and monitor your electrolytes and labs. Blocking NMDA receptors restarts metabolism, but we don’t have all the co-factors ready and I’m worried that fragile people need close monitoring. There is a risk that you don’t have all the cofactors for finishing the conversion from pyruvate to acetyl CoA. I think that this can lead to mitochondrial aldehyde toxicity. I sent a message to Dr Marrs about this. She plans to look at what I wrote on Monday.
I think whenever we’re using an NMDA blocker, UNDER SUPERVISION OF A HEALTHCARE PRACTITIONER we have to do a very gentle diet where we consistently eat only a small amount of carbohydrates every 2 to 2.5 hours so that we don’t go into full-blown lactic acidosis because lactic acidosis will get us stuck in the same pathway. Lactic acidosis could lead to the same increase in the CDO activity that I described in the video. So, it would be a low carbohydrate diet but not carbohydrate-free because to avoid gluconeogenesis as making glucose uses GTP. GTP is needed for making MPT and molybdenum cofactor.
Here is a link to the video. Here is a link to a HYPOTHESISpaper.
how I think that vitamin A metabolism issues result in SIBO or SIFO. I hope to have some diagrams at some point, but here is my general idea of what is happening. I think the development of SIBO/SIFO is multifactorial, but I will explain it from a vitamin A point of view.
In general vitamin A is needed at ***physiological levels*** in the gut to fight both candida and also bacteria. Zoey had high serum A which the GI doctor, me (dietitian), and the GI dietitian assumed was an accumulation of toxic amounts of vitamin A from some unknown alteration in enzyme activity perhaps. She is very unique. So, the solution that we all agreed on was ZERO vitamin A including no eggs (I didn’t know that Dr. Smith or Grant G existed at that time, so this was all on my own initiative).
When she went low vitamin A, but had high serum A, all parties assumed that she would have plenty of vitamin A to use in her GI tract. What we didn’t account for is that although vitamin A bound to RBP4 can get back to the GI tract, it still has to be taken up and stored in cellular retinol binding protein. Also, it has to become retinoic acid in those immune cells that take it up.
****The reasons why I think dietary vitamin A has helped her is because the enterocyte can metabolize vitamin A into retinoic acid using ALDH1A1. This retinoic acid can then transfuse into the blood and gut associated lymphoid tissue to be used by the immune cells. It bypasses the need for retention of retinol inside of cells of the immune system****
Zoey was struggling with both activities it seems. First, she struggles with calcium dysregulation. Zoey struggles with a mild form of renal tubular acidosis due to a down regulated mRNA expression for a bicarbonate transporter, but also, she has an underlying issue with sleep apnea which induces a pathway called hypoxia inducible factor 1 alpha (HIF-1alpha). This pathway causes lactic acidosis. The acidosis causes loss of magnesium in the urine. Magnesium is crucial for controlling calcium levels in cells by being part of the EF hand of a calcium binding molecule calmodulin that regulates cellular dynamics. Calmodulin controls the STRA6 pore that decides on whether vitamin A stays in the cell or leaves the cell. When cellular calcium is high, vitamin A is preferentially effluxed back into the blood onto RPB4 leaving the cell void of retinol. This includes immune cells as macrophages, B-cell, and mature dendritic cells all contain STRA6.
In macrophages, just binding of RPB4 to TLR4 induces an inflammatory response. The increase in NF-KB that happens when RPB4 docs with macrophages upregulates HIF-1alpha gene transcription. (LPS can also induce the NF-KB pathway in macrophages leading to HIF-1alph activation.) This should help increase the ability of immune cells to fight fungal infections. However, because of calcium dysregulation, retinol doesn’t stay in the cells to be able to make retinoic acid which upregulates dectin-1 receptor production. Neutrophils and dendritic cells also respond to retinoic acid by increasing the dectin-1 receptor.
So, you may have an increase in HIF-1alpha pathway (which by the way causes lactic acidosis which worsens magnesium losses and increases ionized calcium as well as increases calcium uptake into cells), but a decreased recognition of candida. Candida is recognized by the immune system specifically through the fact that it has beta-glucan on its cell wall. The immune system recognizes beta-glucan using the dectin-1 receptor. If dectin-1 expression is low, this can result in decreased ability to fight fungus despite having HIF-1alpha turned on. I don’t think the fight is completely stopped though. It’s just less efficient. Retinoic acid upregulates dectin-1 receptor. It isn’t necessarily absent when retinoic acid is low. However, in B-lymphocytes, lack of retinoic means decreased secretory IgA for binding to LPS. Less retinoic acid also means poor conversion of monocytes to macrophages and failure to recruit more white blood cells from the blood to the sight of infection in the gut due to less cytokine production. When LPS increase, it also triggers macrophages to make NF-KB which can increase HIF-1alpha transcription and lead to the HIF-1alpha pathway. The only gut bug that can dampen this response is enterohemorrhagic E. coli. And that is NOT a good thing. Either having EHEC or having poor retinoic acid production will cause candida to flair badly. This is what leads to SIFO I believe.
Lack of sulfate causing damage to immune cells in the gut because of anhydroretinol toxicity?
Another factor for Zoey that I see happening in many people is the lack of sulfate for back up alcohol metabolism in the gut. Specifically, b-lymphocytes have been shown to produce anhydroretinol (alcohol + acetic acid at human pH = anhydroretinol). Excess alcohol in the gut can result in death of B-lymphocytes which means less secretory IgA and more risk for LPS entering the body.
This lack of sulfate was coming from two issues. 1: oxalate (uses same transporter as sulfate) 2. Molybdenum deficiency (needed for SUOX enzyme to turn sulfite into sulfate). The oxalate issue was because a well meaning dietitian’s handout (kidney dietitian .org) has plantain as low oxalate when it is not. The molybdenum deficiency was caused by high amounts of vitamin A return to the liver I think and when vitamin A is high, the body using AOX for metabolism. Also, though, due to chronic hypoxia, Zoey had been stuck in the HIF-1alpha pathway that is the same pathway used to fight candida. In this pathway the enzyme CDO is upregulated which increases the need for SUOX activity. However, on top of that sleep apnea increase xanthine oxidase activity which also needs molybdenum. And then to add to that mess, Zoey has upregulated mocosulfurase mRNA expression which means she preferentially makes AOX and XO over SUOX. Phew. So in general she became very slow at SUOX, but needed more SUOX due to HIF-1alpha upregulation of CDO. (Jenny Jones, PhD and I have done significant work on this Moco Steal pathway. I will share more about it soon.)
Here is where SIBO comes in…
One of the things that HIF-1alpha does when fighting candida is increase the gene transcription for heme oxygenase 2 (HO-2). HO increases carbon monoxide levels which leads to a down regulation of CBS enzyme activity. This means LESS hydrogen sulfide. So, to compensate, the body might allow H2S producing bacteria to grow to help the body out with the lack of H2S available due to high RBP4 or LPS triggering NF-KB which increase HIf-1alpha. (Something to think about is that high serum A is a sign of high RBP4, but low serum A is NOT a sign of low RBP4 as RBP4 doesn’t have to be carrying vitamin A. It can carry palmitate and also anhydroretinol. Vitamin A can be low on tests and there still be an issue with RBP4).
This theory of growing sulfur metabolizing bacteria is actually a Greg Nigh theory of SIBO, but the process of what is causing us to not make enough at times is something I think goes along with vitamin A dysregulation. I love Greg’s theory, and it fits with what Zoey experienced with vitamin A dysregulation. It fits 100% with her developing SIBO when going very low vitamin A.
Other inducers of HIF-1alph besides hypoxia and candida include probably spike protein, nicotine, and beta-glucan.
Can HIF-1alpha pathway cause Vitamin A dysregulation and high serum vitamin A levels? Yes. I think it can.
Now back to that upregulated HO-2 enzyme from the HIF-1alpha pathway. As CO levels increase, O2 levels in the tissue drop. This eventually causes slowing of HO-2, and so then CBS turns back on because there is less carbon monoxide. When this surge of H2S happens, potassium leaving the cell is blocked. This causes an uptake of calcium and high intracellular calcium levels if the PMCA pump is not working well (calmodulin dependent and also needs magnesium on the EF hand). This is when I think there is even more vitamin A dysregulation as vitamin A effluxes back out of cells and worsens the whole situation especially for immune cells in the gut that require retinoic acid to make secretory IgA.
So there are many places in this path to think “what comes first, the chicken or the egg”. Is it high serum A that causes all the dysfunction? Is it the poor production of retinoic acid in the gut? Is it hypoxia alone inducing the HIF-1alpha pathway? Is it sneaky anhydroretinol? Maybe using a cod liver oil brand with a bunch of anhydroretinol?
What has helped for Zoey?
Electrolyte balance. This is difficult to translate to all people. I suggest tracking your intakes in Cronometer and making sure you get at least 2400 mg sodium (need the Na-Ca exchange transporter working). Goal for potassium is what your RDA is when you put in your stats. Then on magnesium, if you are in acidosis, making sure to get twice the RDA perhaps. I don’t go higher because too much magnesium can act like a beta blocker and slow down the gut more. I typically don’t push electrolytes in doses. I add them to 2 liters of water and give over the course of one day. I use a combination of citrate, gluconate, and bicarbonate. I sort of just have found what works with her. I keep the bicarbonate at no more than 250 mg of potassium bicarbonate per day because going higher seems to make candida grow. Plus, huge shifts in bicarbs sends her reeling because even though she has RTA most of her acidosis seems to be lactic acidosis or glycolate poisoning from hypoxia.
Zoey also gets 250 to 500 mcg of B12 per day added to the 2 liters of water. She gets 250 to 350 mg betaine as well. Both of these help with the iNOS causing high nitric oxide and a functional B6 deficiency
She gets 15 mg of nicotinamide riboside at each meal that has 25% of the daily value of vitamin A (usually 1 to 2 meals per day – goal is 50% daily value). We don’t use nicotinic acid because it causes calcium dysregulation to worsen. We don’t do high dose because it puts burden on AOX which means more of MoCo steal and worsening sulfite issues. I don’t recommend any niacinamide, nicotinic acid, or nicotinamide riboside to anyone who is a carrier of enterohemorrhagic E. coli as it has been shown to encourage the adherence of this E. coli to the gut. I am thinking about switching to NMN, actually.
She gets 100 mg ginger extract at each meal with vitamin A to increase the ALDH1A1 enzyme activity in the gut, so she makes retinoic acid. She is thankful to have a g-tube for this reason! Haha (yes, she does eat orally, but gets liquids through a stomach tube)
We use Orthomolecular Motility Pro as a prokinetic agent. It works well. I give it only once a day on an empty stomach.
We keep fat intake to 10-15 grams per meal maximum to lower LPS entrance in chylomicrons. This also helps to decrease TMAO production by bacteria in the gut. The higher the fat, the more TMAO. That leads to lower bile acid pools and worsening constipation. Constipation is not good for SIBO and SIFO.
What if periodic “oxalate dumping”, intolerance to dietary oxalate, and subsequent low NAD/NADH ratio leading to hypervitaminosis A (retinol/retinaldehyde toxicity with simultaneous retinoic acid deficiency) is caused by a block at pyruvate dehydrogenase complex (PDHC) due to sleep apnea or due to the immune system activation during a fight against fungal infections or bacterial infections? What if lack of oxalobacter formigenes (breaks down oxalate) isn’t the only issue?What if gut dysbiosis triggers our own body to produce oxalic acid?
***I’m a dietitian and not a doctor. This post is written for informational purposes only and not meant as medical advice. Any therapeutic suggestions made in this post should be discussed with your own personal health care provider prior to making any changes to your diet, nutrition, supplements, or lifestyle.***
On August 6th, 2023 I wrote this article Oxalate: A Potential Contributor to Hypervitaminosis A – Hormones Matter for hormone matter regarding a hypothesis that hyperoxaluria and high dietary oxalate contributes to hypervitaminosis A by altering cellular NAD to NADH ratio contributing to a high retinol state and a low retinoic acid state. I felt like dietary oxalate was a huge component to Zoey’s chronic hyperoxaluria and do believe that plantain flour, recommended by a dietitian who is an expert in kidney disease, was the cause of Zoey’s unfortunate oxalate poisoning. However, what if the reason that Zoey is unable to tolerate dietary oxalate is actually that she, herself, is an over producer of oxalate in her body depending on her oxygen levels or immune function?
Hypothesis: The cause of new onset idiopathic hyperoxaluria and glycolate toxicity may actually be overactivation of HIF-1alpha due to fighting bacteria, fungus, hypoxic conditions, and/or sleep apnea.
My daughter Zoey is unable to tolerate more than 50 mg of oxalate a day and is sick every afternoon with headache and nausea. Why? Although she lacks the genetics for primary hyperoxaluria, she periodically becomes an oxalate producer that at first seemed random. She also struggles with high glycolate levels on her organic acid test despite doing all the right things recommended for resolution of glycolate toxicity. Looking back on her twelve years of life, I have realized that her bouts of high urinary oxalate levels and horrible headaches weren’t random at all, but occurred when she was fighting a bacterial or fungal infection or when she was non-compliant with her CPAP mask for sleep apnea.
Recently this all worsened, and many of my side clients were experiencing similar issues. This prompted me to find an answer to this mystery. I asked myself, “What if hyperoxaluria, despite consuming a low oxalate diet, and having no genetic aberrations to cause hyperoxaluria, isn’t the product of bad glycine metabolism? What if it is the after effect or the ongoing effect of the activation of HIF-1alpha, a factor that is made in the body whenever it is fighting fungus or bacteria or when experiencing hypoxia?”
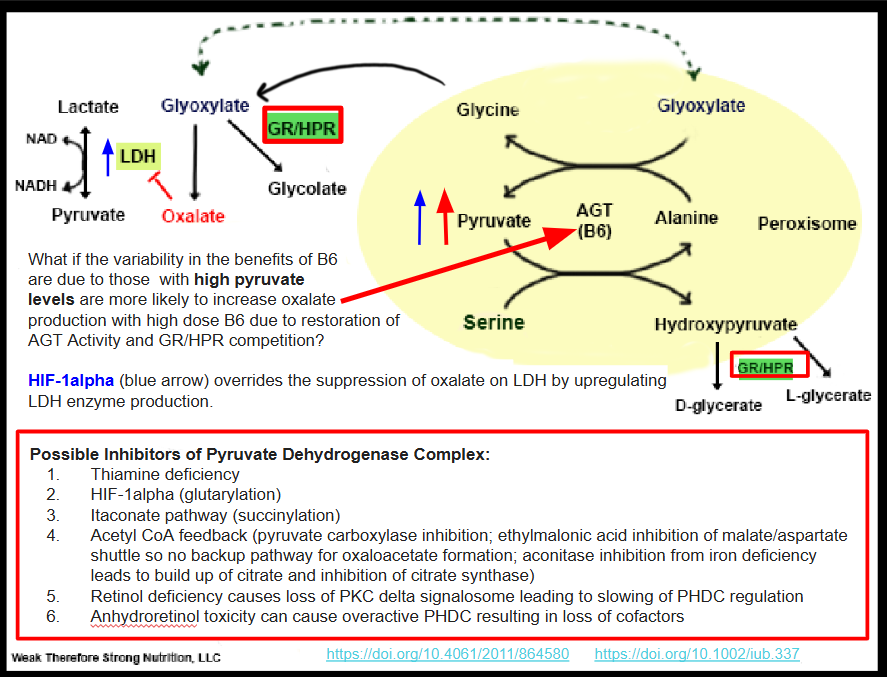
HIF-1alpha causes a block at pyruvate dehydrogenase complex (PDHC). After extensive research and comparing of organic acid test results among those who share them with me, I believe that a block at PDHC by HIF-1alpha may be the number one contributing factor to production of endogenous oxalate in the setting of B6 sufficiency, and this block at PDHC can account for a paradoxical reaction to B6 supplementation increasing oxalate formation. In fact, when people are functionally or marginally deficient to severely deficient in B6 I propose that they will make less oxalate while producing more glycolate, which unfortunately poisons them if the kidneys cannot keep up with excretion of this metabolite.
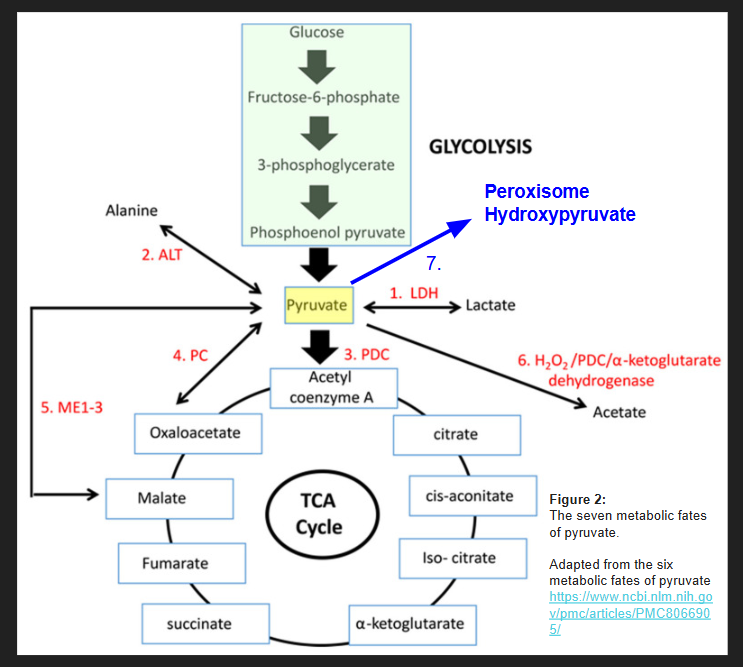
HOW DOES A BUILD UP OF PYRUVATE CAUSE HYPEROXALURIA?
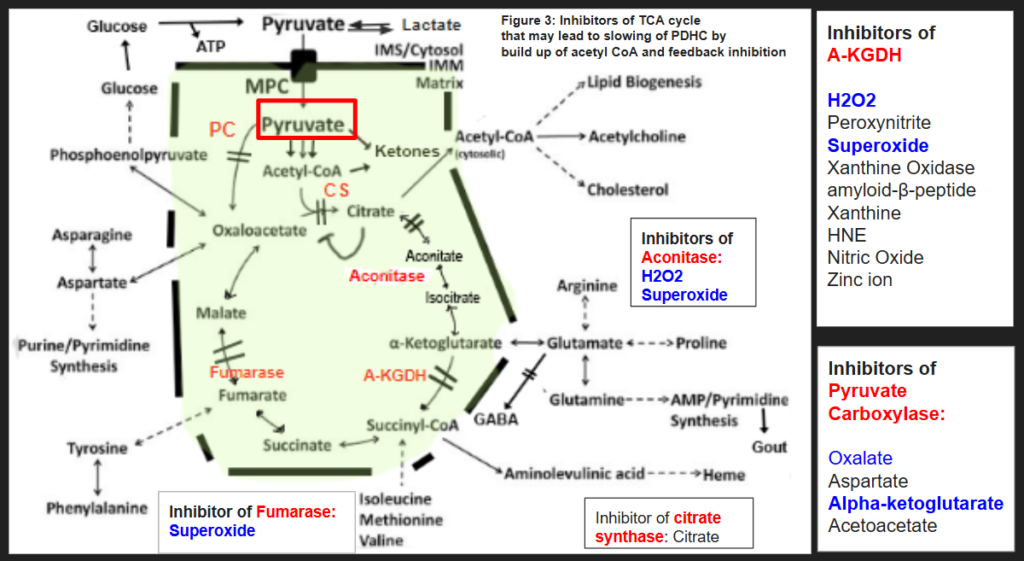
Pyruvate in excess that is unable to escape by six different metabolic pathways (explained further below) is shuttled to the peroxisome and combines with serine via a reaction metabolized by alanine glyoxylate transferase (AGT) to hydroxypyruvate (Figure 1). Hydroxypyruvate is then metabolized by Glyoxylate Reductase/Hydroxypyruvate Reductase (GR/HPR) to D-glycerate and L-glycerate. This enzyme is the same enzyme that metabolizes glyoxylate to glycolate. If this enzyme is tied up in hydroxypyruvate metabolism due to excess pyruvate build up, it may not metabolize glyoxylate to glycolate. The end result is more glyoxylate is made into oxalate via the enzyme lactate dehydrogenase (LDH). This should have feedback inhibition on LDH and stop the formation of further oxalates. However, if someone is fighting a bacterial or fungal infection, or even has excessive inflammatory macrophages produces excess NF-KB such as in the acute phase response to illness, HIF-1alpha increases. HIF-1alpha, in turn, upregulates the enzyme LDHA and so feedback inhibition by oxalate is overridden by the sheer abundance of lactate dehydrogenase available. LDHA is the same enzyme isoenzyme involved in glyoxylate to oxalate conversion and is actually a therapeutic target for individuals with primary hyperoxaluria. See Figure 1 for a pictorial demonstration of this pathway and a summary of inhibitors of PDHC.
When I added B6 to my own family members regimen to stop oxalate production, they only worsened. Of course, B6 is necessary for many reactions in the body, but what if the worsening of oxalate formation from adding B6 is due to supporting the already overworking AGT enzyme and the tying up of the moonlighting GR/HPR enzyme in dealing with hydroxypyruvate? Perhaps dealing with the block in pyruvate metabolism is the better way to solve the problem?
HYPEROXALURIA DUE TO EXTENSION OF IMMUNE FUNCTION
Whether fighting a bacterial or fungal infection, HIF-1alpha gets involved in redirecting glucose metabolism to provide energy for T-cells of the immune system via alteration of metabolism including inhibition of PHDC. In the research area myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) community, Dr. Rob Phair working with Dr. Ron Davis have a theory that ME/CFS occurs when cells other than the immune system take on an immune pathway. These cells adopt the itaconate pathway. Dr. Fair’s Itaconate Pathway theory for ME/CFS makes sense from a clinical standpoint as more and more people are experiencing altered metabolism.
If cells outside of the immune system are now taking on immune function, and there is an increase in HIF-1alpha orchestrating a multi-cellular response in the liver and kidneys, then it’s possible that HIF-1alpha pathway itself becomes a source of excess pyruvate that can contribute to hydroxypyruvate competition for GR/HRP. In addition, HIF-1alpha itself is tagged with hydroxyproline for degradation by proteasomes, and hydroxyproline can be a source of oxalate formation in PH type 1, 2 and 3 according to this in vivo human study. Overall, it appears that the adoption of a HIF-1alpha change in energy metabolism could be a major contributor to hyperoxaluria. My own daughter has experienced hyperoxaluria without having any genetic discrepancies as a contributing factor. What she does have, though, is chronic candida, and chronic bacterial infections, as well as sleep apnea as contributors to overactivation of HIF-1alpha.
WHAT ACTUALLY IS BLOCKING PYRUVATE METABOLISM?
HIF-1alpha itself has been shown to block PDHC leading to a buildup of pyruvate. However, it isn’t the only cause of a block at PDHC. In figure 1, I list the possible blocks of PDHC. Of course, thiamine deficiency is an obvious cause of loss of PDHC function and Dr. Londsdale and Dr. Marrs are the authorities on the downstream effects of thiamine deficiency. Yet, there are some people not responding to thiamine supplementation. These are the outliers. These are a few of the people in my family and some of my clients who do not improve with high dose thiamine, and possibly some of the people that I interact with online in forums who aren’t responding to thiamine supplementation. It may be that although thiamine can definitely fix the PDHC complex, along with other cofactors, it may or may not fix a metabolic block caused by the immune system or hypoxia. One question to explore would be, “If thiamine over-rides a metabolic block produced by the immune system, will bacteria and fungus be successfully eradicated?”
The lack of improvement with supplemental thiamine may be because HIF-1alpha causes a block at the enzyme pyruvate dehydrogenase complex (PDHC) leading to a buildup of pyruvate that must be shuttled somewhere. Figure 2, adapted from the six metabolic fates of pyruvate shows various ways that the cell deals with excess pyruvate. The seventh possible place for pyruvate to be shuttled is the peroxisome to form hydroxypyruvate as I show in Figure 1.
So, it’s possible that as long as there aren’t any blocks in enzyme #1-6 shown in figure one, that a person will not actually push pyruvate to the peroxisome to make hydroxypyruvate despite a block at PDHC for various reasons. Hyperoxaluria from a block at PHDC only becomes a problem when these other escape pathways for pyruvate are shut down, or when lactate is unable to be cleared from the body due to uremia (such as that occurs in chronic kidney disease or an obstructed ureter that occurs in kidney stone formation).
DIVERSION OF PYRUVATE AWAY FROM THE PEROXISOME
I think the goal of someone with hyperoxaluria who is stuck in a chronic state of fighting infection or struggling with hypoxia, is to concentrate on the six metabolic fates of pyruvate below. In figure 3, I have summarized some of the blocks of the enzymes of the TCA cycle that can lead to inhibition of pyruvate carboxylase and citrate synthase. Feel free to discuss these blocks with your own health care provider as I am unaware of your medical history. I hope to make a few short videos about how to get around each block in the pathways below in the future.
ORGANIC ACID TEST PROOF THAT BLOCKS AT PDHC CAUSE HYPEROXALURIA
Although I am currently taking a break from full time practice to care for my daughter, I often glance at organic acid tests for people who contact me and help them on the side, especially when someone has lost all hope. I have seen this trend of hyperoxaluria or high glycolate many times over in organic acid testing when someone is struggling with fighting a bacterial or fungal infection and has had to mount what seems to be a whole-body response. Here is a link to a recent YouTube video I have made that has screenshots of organic acid testing and explanations of how these results fit the picture of alterations in pyruvate metabolism contributing to hyperoxaluria. The video includes a little bit of theory on a sneaky metabolite of vitamin A, anhydroretinol, that could be causing some people to develop thiamine deficiency and also immune dysfunction leading to chronic infections that trigger chronic activation of HIF-1alpha.
HIF-1ALPHA DEGRADATION IN NORMOXIA CAUSES HYPEROXALURIA AND GLYCOLATE POISONING.
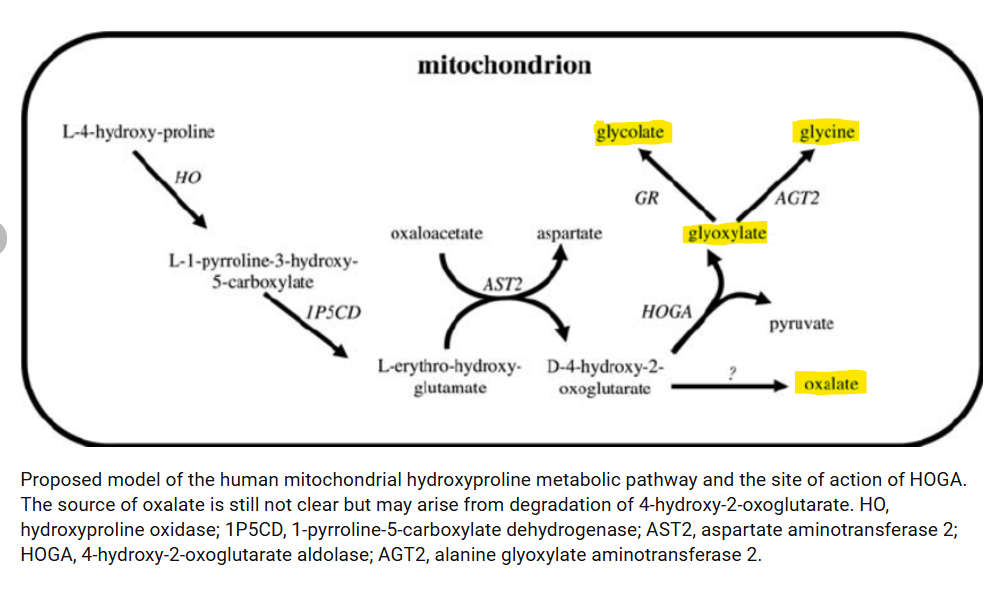
During a fungal infection in the absence of hypoxia, HIF-1alpha degradation will lead to increasing levels of hydroxyproline after it is ubiquitinated and degraded. This leads to an excess of hydroxyproline that must be metabolized as it is unable to be used again to synthesize proteins. This can lead to high oxalate formation as well as glycolate formation. This process can also happen in individuals with sleep apnea that can maintain normal oxygen levels while awake. These individuals cycle between low oxalate and glycolate states to high oxalate and glycolate states. In fact, Zoey, my daughter with sleep apnea, consistently has a headache and nausea every afternoon no matter what foods I feed her. She wakes up a bit sleepy, has a pretty good morning, and then goes downhill by the afternoon and requires Zofran for nausea and lays on the couch completely incapacitated. I believe this could be due to her body catching up with HIF-1alpha degradation with the resulting production of glycolate and oxalate. It appears she isn’t alone. Hydroxyproline degradation has been found to be a contributor to oxalate formation in all three types of primary hyperoxaluria. Perhaps the source of the excess hydroxyproline is overactivation of the HIF-1alpha pathway. Figure 4 shows that hydroxyproline is metabolized in the mitochondria to glyoxylate, glycolate, and glycine, and oxalate.
EVIDENCE THAT THIS IS TRULY HAPPENING? ORGANIC ACID TESTS FOUND IN VIDEO BELOW.
The video below goes through individual labs, and, in particular, shows one client who ate carnivore diet for about one week before her second organic acid test. This diet completely resolved her hyperoxaluria and high glycolate levels! This particular client’s information is at about 27 minutes and shows that changing to a very low carbohydrate diet alleviating her hyperoxaluria.
SOLUTIONS TO HIF-1ALPHA INDUCED HYPEROXALURIA?
Honestly, I have thought long and hard about this. My daughter Zoey has moderate to severe obstructive sleep apnea causing chronic hypoxia and induction of HIF-1alpha. I explored the possibility of inhibiting the gene transcription for HIF-1alpha with known inhibitors. I think for my own daughter, Zoey, I will not be trying to inhibit HIF-1alpha through use of known inhibitors. I believe that this could be counterproductive if she is fighting a fungal or bacterial infection.
However, I do plan on controlling her sleep apnea issues. She wears CPAP at night but is quite non-compliant with it. Her sleep medicine doctor is willing to try a little bit of oxygen, but we are also doing an evaluation with an ENT to make sure that her sinus passages are normal as well as an evaluation with neurosurgery because she has an alteration in her C1 vertebrae that could be causing the hypotonic sleep apnea. I highly recommend if someone has an underlying apnea issue, to have a CT scan of the sinus and C1 vertebrae. These areas, if altered, can be a major source of hypoxia.
Something else to consider is that fighting off pathogens causing high levels of reactive oxygen species (ROS) through NOX, but also sleep apnea itself induces the activity of an enzyme, xanthine oxidase, that also creates reactive oxygen species which inhibits proline hydroxylation of the pro-oxidative stress, HIF-1alpha. This keeps HIF-1alpha in an active state causing massive oxidative stress through NOX and down regulation of antioxidant HIF-2alpha. This type of high activity of NOX is pathogenic and can lead to excessive ROS and disease. How would someone know if they have elevated XO activity? They could monitor uric acid urine levels while eating carbohydrates and on a low purine diet (as purines will definitely increase uric acid alone). Xanthine oxidase converts xanthine to uric acid. Inhibitors of xanthine oxidase might be beneficial for people with chronic hypoxia to lower the overall oxidative stress on the body. Although drugs are available for this purpose, you might choose natural inhibitors of xanthine oxidase with your own healthcare provider.
Another goal that I have for my daughter, Zoey, is to avoid fungal and bacterial infections. This, of course, takes a bit more work from a nutrition standpoint. Definitely seek out a practitioner who understands the importance of nutrition for immune health as avoiding chronic fungal or bacterial infections is likely the key to prevention of extreme alterations in metabolism by HIF-1alpha expression. However, I do think having normal gene transcription for HIF-1alpha is crucial to mount a defense against pathogens.
Something I definitely don’t recommend if you have an active bacterial or fungal infection is a carnivore or keto diet long term. These diets may help with working around the block in metabolism at PDHC by lowering total glucose availability, but if there is truly an infection that needs to be resolved, a carbohydrate free diet will starve immune cells of very much needed glucose. Glucose is needed to fuel the HIF-1alpha upregulated pentose phosphate pathway to make NADPH which is used by NADPH dependent oxidase (NOX) to produce superoxide for killing pathogens. When I noticed that someone I am caring for has high microbial or fungal markers on their OAT, even though they might feel better on a keto diet because it lowers total pyruvate levels and decreases both glycolate and oxalate production, I think it would be better to continue to eat a well-balanced diet to fuel the immune system. They might realize that eating about 100 grams of carbohydrate per day is a happy medium where they don’t feel sick from glycolate and oxalate, but they are still able to maintain immune function. This would be per person of course.
Overall, I think discovery of this possible mechanism of increased oxalate and glycolate formation due to a block at PDHC from HIF-1alpha is going to change my daughter’s life. We have a new focus on how to promote her health and well being. The mystery of hyperoxaluria is not so much a mystery afterall.
The importance of ALDH7A1 competition in anhydroretinol formation
ALDH7A1 is an enzyme that moonlights throughout the cell and goes where it is needed based on cellular metabolism. In oxygen deprivation it becomes a regulator of cellular energy homeostasis. This enzyme also is tied up in lipid peroxidation detox, histamine metabolism, and betaine production in the setting of a low betaine diet (Gluten free diets are a huge cause of burden on ALDH7A1). Betaine is the cofactor for BHMT that runs methionine salvage pathways in the liver, kidneys, and small parts of the brain (this is found in a model of MS) while under conditions of low 5-methylfolate production or B12 deficiency or during a high nitric oxide state which inhibits the enzyme methionine synthase.
If ALDH7A1 is not doing lysine metabolism this results in an increase in P6C and inactivation of P5P (vitamin B6). This can lead to insufficiency of B6 and the body will have to choose where to use it. One problem with low B6 levels is that this leads to poor denovo NAD production. NAD is a cofactor needed for ADH that is required to deal with the tiny bit of alcohol produced during digestion (3 grams – about 1/2 shot glass) and also the alcohol being made from gut dysbiosis (alcohol producers such as candida, aspergillus, s. boulardii. s cerevisiae, e.coli, clostridia).
If you can minimize lipid peroxidation, the need to make betaine, or consume enough betaine, and limit histamine needing ALDH7A1, then this will allow normal lysine metabolism and normal de novo NAD metabolism and then we are no longer reluctant alcoholics.
This requires focusing on each person’s ability to deal with oxidative stress as well as their methylation pathway. The innate immune system plays a role as well because activated macrophages cause increased iNOS expression which results in increased nitric oxide formation and inhibition of methionine synthase. Thus, chronic bacterial infections will trigger a major downstream effect resulting in a risk factor for making anhydroretinol.
Anhydroretinol obliterates f-actin which is a cytoskeletal protein. This scaffold controls the calcium exporter PMCA that is found in all cells as a housekeeper to regulate calcium. Actin is found in the G-Actin state or the F-actin state or an in-between state called an oligomer. When it’s an oligomer it turns on calcium export to bring the cells back to a normal intracellular calcium level, so it doesn’t go through apoptosis. When it grows into F-actin, PMCA is turned off and calcium accumulates. It cycles back and forth between the long F-actin chains and oligomers, but being destroyed down to G-Actin by anhydroretinol would in theory cause loss of PMCA function. If anhydroretinol stays around too long, then apoptosis occurs due to loss of cytoskeleton and hypercalcemia.
If cellular levels of anhydroretinol are not to the point of causing apoptosis, the resulting excess calcium in cells will cause overactivation of calmodulin. Calmodulin is what causes the Stra6 pore to prefer to stay open, but instead of doing a normal influx and efflux, when calcium is high it lets retinol leave the cell and doesn’t let retinol in cells as much. This also occurs in the setting of magnesium deficiency because calmodulin overactivation is controlled by an EF-hand on calmodulin that requires magnesium. If calmodulin is overactivated, this causes low retinol uptake and thus cellular retinoic acid deficiency, but high serum vitamin A levels due to efflux back onto RBP4 which we classically see as vitamin A toxicity. What I have found is that vitamin A toxicity is actually both a toxicity and deficiency at the same time.
The provision of “fresh” retinol in a cell from postprandial chylomicrons could save the day because it has been shown to stabilize F-actin in a model where anhydroretinol destroys it. Dietary retinol could work as long as alcohol is dealt with properly in the gut, and the person does not make anhydroretinol in their gut. Anhydroretinol can be made into rehydroretinol when cellular alcohol levels are low, but it is only about 7% of the activity of the original retinol molecule, and so whether or not it also can act to stabilize F-actin is questionable.
One more interesting aspect of F-actin is that it is the scaffold for KEAP1 which regulates NFR2 activation. If F-actin is destroyed by anhydroretinol, this causes alteration in KEAP1 scaffolding and this could lead to dysregulation of NRF2. I’m finding that mostly it leads to overactivation of NRF2 which causes hyperkeratosis as NRF2 not only turns on genes for dealing with oxidative stress, but also turns on keratin producing genes. I believe this is the cause of hyperkeratosis. My vitamin A toxic client experience shows they are all struggling from hyperkeratosis and keratosis pilaris.
How do we keep anhydroretinol in control? We control alcohol levels.
What factors deal with alcohol. 1. NAD (The biggest concern is de novo production via kynurenine pathway or maximize recycling of cytosolic NAD via avoidance of high oxalate diet if needed.) 2. SULT (acts as a backup buffer) (SULT1A1 in human intestines) 3. Vitamin E (can help restore NAD recycling and prevent lipid peroxidation)
4. CYP2E1 (although this is the last resort because it causes oxidative stress)
What can lower #1 NAD? 1. Functional B6 deficiency from ALDH7A1 competition leads to low NAD production 2. Cytosolic recycling of NAD is inhibited by oxalate inhibition of LDH 3. Pyroluria 4. High alcohol intake
5. Gut dysbiosis
What can lower #2 SULT? 1. SUOX issues (Moco, B2, B6) 2. Arsenic, chlorate, and perchlorate inhibit PAPS synthase 3. Salicylates inhibiting SULT 4. Quercetin inhibiting SULT 5. Excess H202 causing sulfite radicals instead of sulfate to produce which causes lower levels of PAPS
What can cause issues with #3 vitamin E? 1. Low E diet due to eliminating too many foods 2. Excess oxidative stress causing increasing vitamin E needs
What can cause issues with #4 CYP2E1?
Inhibitors (which could be good due to CYP2E1 causes more oxidative stress, but if #1-3 is down, CYP2E1 may be the only way to deal with alcohol in the liver). Inhibitors of CYP2E1 include – quercetin, cannabidiol, niacin, niacinamide, menadione
Calcium excess in cells has side effects (here are just some of them):
1. Seizures
2. Mast cell activation
3. Decreased aromatase activity leading to low estrogen and slow PEMT needed to make PC for making CDP-Choline for plasmalogens (leads to membrane instability)
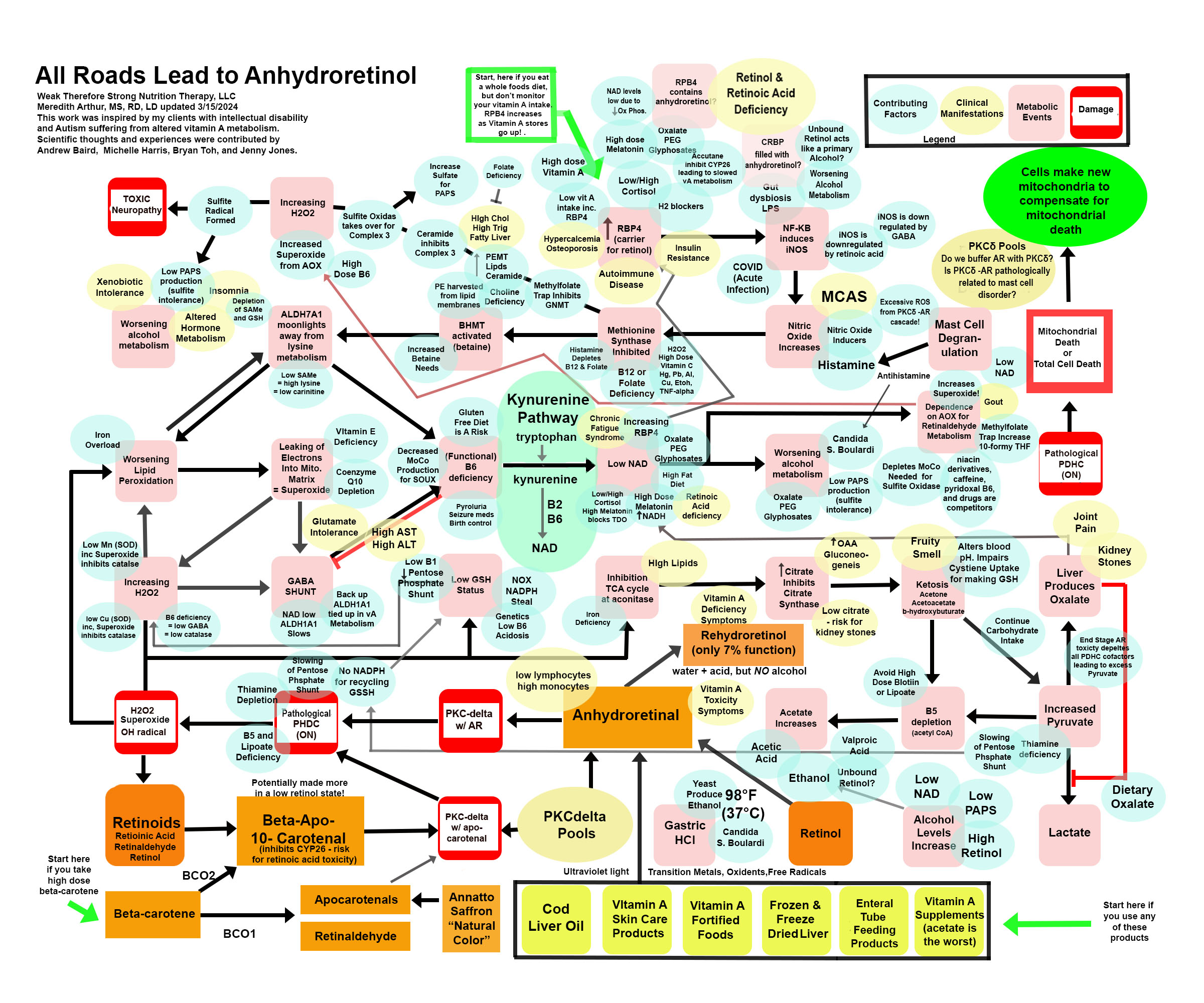
Anhydroretinol (AR) is a metabolic byproduct of vitamin A degradation as well as vitamin A metabolism. We can consume it in varying amounts in our diet, and our body, in the absence of a primary alcohol, will convert AR to rehydroretinol using acid and water. Rehydroretinol only has 7% of the function of retinol. However, when alcohol levels are high due to candida, low production of PAPS (sulfate deficiency) to buffer alcohol we make in metabolism, or alcohol intake, then the normal acid we make in metabolism in small amounts (or in large amounts if we are pantothenic acid deficient), acetic acid, becomes a catalyst to make AR. I’ll be posting more about this metabolite of vitamin A because I believe this is THE SNEAKY version of vitamin A that causes “vitamin A toxicity.” Below is a “road map” of anhydroretinol. You can start at the green arrows and work your way through the map, you can look at the blue ovals which are contributing factors that can lead you into this pathway, or you can look at the yellow ovals to find symptoms or lab work that matches your own health issues. This is a work in progress and some is hypothetical, but what I am sure of is the damaging effects of AR on the individuals I care for is directly related to the pathological damage it does to energy metabolism.
If you would prefer to listen to me babble on and on about this topic, here is a video! Enjoy. Or just scroll on by and read to avoid my crazy rant about AR. Hahaha!
(At the end of this video there is a link to a correction video to show that beta-apo-10′- carotenol is NOT the carotenal that inhibits CYP26. This is actually beta-apo-13′-carotenone. It’s made from retinol, retinaldehyde, and retinoic acid during oxidative stress. It is an inhibitor of CYP26 and can cause accumulation of retinoic acid leading to feedback inhibition of RALDH. This will cause accumulation of retinaldehyde and possibly formation of A2E. Also, beta-apo-13′- carotene is an antagonist for specific RXR and can inhibit gene transcription. It seems to be not a good guy. It’s the reason why a low vitamin A diet that still provides retinoic acid from animal protein, could lead to a worsening health status over time. Beta-apo-10′-carotenol is made from BCO2 metabolism, and if you have a slow BCO1 enzyme, this is still a risk if it is not buffered with retinol. It can act in place of retinol in PKCdelta and pathologically turn on PDHC. )
So…I’m a dietitian, not a doctor. This blog isn’t medical advice, but only a guideline to share with your own personal healthcare provider. It isn’t intended to diagnose or treat a condition without supervision of your own health care practitioner. Please don’t make any changes to your diet, supplements, nutrition, or medications without talking with your own provider who knows you and your health needs.
That being said, please read on….it’s intriguing!
Why is AR so toxic?
AR’s toxicity is due to quantum mechanics. Yes, that’s right. I am not a quantum mechanics expert. I’m a dietitian, but the general gist of the problem is that when retinol becomes AR, it shifts the double bond orientation, and it no longer behaves like retinol.
Retinol plays a pivotal role in a signaling molecule called PKCδ signalosome. This signalosome consists of PKCδ, retinol, and cytochrome C of the electron transport chain. The PKCδ signalosome works to sense the energy level in the cell. When the cell doesn’t need energy anymore, PKCδ inhibits pyruvate dehydrogenase kinase 2 (PDK2). PDK2 usually removes a phosphate groups from Thiamine Pyrophosphate on E1 to make the enzyme pyruvate dehydrogenase complex (PDHC) inactive. PDHC converts pyruvate to acetyl CoA. Turning off PDHC helps to stop the influx of acetyl CoA into Krebs cycle and keeps the cell from creating too many electrons in the intermembrane space.
This regulation is crucial because if too many electrons accumulate, then they leak back through the mitochondrial membrane into the matrix and form superoxide. Superoxide causes a metabolic cascade that shuts down the Krebs cycle at the level of alpha-ketoglutarate. Alpha-ketoglutarate is exported from the mitochondria to the cytosol where GABA is made. GABA is a sort of “antioxidant” for the liver because it upregulates enzymes of that deal with reactive oxygen species. Typically this restores the mitochondria to normal, but when individuals have broken pathways involved in this mopping up of reactive oxygen species, they struggle with the damages that occur.
The problem with AR taking the place of retinol in the PKCδ signalosome is that AR pathologically turns on PDHC. It doesn’t shut off. This eventually leads to depletion of all the cofactors needed to support restoration of the cells redox balance. In addition to AR causing this issue, apo-carotenal can also take the place of retinol on PKCδ signalosome and cause the same issue. The authors of the quantum chemistry study on retinoids report that if there is enough fresh retinol in the cell to “buffer” AR or apo-carotenal, then the cell should just have a sort of energy boost with a prolonged “on” state of PDHC, but the question I have is, “Can all people tolerate this prolonged on state or is this the start of an endless pathological cycle leading to more and more AR production within the body in those with broken redox pathways?”
AR toxicity may be due to the fact that it can’t be stored like retinol in hepatic stellate cells.
AR lacks the “-OH” group required for lecithin:retinol acyltransferase (LRAT) reaction using phosphatidylcholine to make a retinyl ester. LRAT is an enzyme that helps to package retinol into cellular retinol binding proteins and also onto retinol binding protein four (RPB4). Our cells have two back up enzymes, acyl-CoA:retinol acyltransferase (ARAT) and Acyl CoA: Diacylglycerol O-acyltransferase 1 (DGAT). These enzymes also require anhydroretinol to have an “-OH” group to interact with their “CoA” to bind a fat to retinol. ARAT and DGAT are back up enzymes for taking care of high levels of retinol in the cell. The acyl CoA portion of the fat they want to add requires pantothenic acid. I believe that AR causes pantothenic acid deficiency by pushing the conversion of pyruvate to acetyl CoA in excess. Interestingly, knockout of DGAT results in mice having alopecia. Many people with vitamin A toxicity experience alopecia. I’ve seen people lose parts of their eyebrows or have patches of hair missing from their heads. If DGAT is tied up in trying to bind retinol or rehydroretinol due to levels are too high in cells (a high NADH state prevents metabolism of retinol and would also prevent metabolism of rehydroretinol), then it’s busy mopping up retinol. That may be the cause of the alopecia, although a functional B6 deficiency is possible as well. The fact that AR can’t be stored, and that alcoholics have been found to have no storage of vitamin A in their liver, makes me highly suspicious that their alcohol intake plus the acetic acid or other possible acids in metabolism pushed them into the process of making AR from retinol.
How do we “detox” AR?
The fact that AR can’t be stored in hepatic stellate cells, means we have to get it out of liver cells. I have a sneaky suspicion that AR is “stored” in PKCδ, but that is yet to be explored as AR research is mostly limited to trying to induce cell death and it’s thought that it’s a very small part of vitamin A metabolism in vivo. However, I suspect AR production is significant in individuals with broken metabolic pathways. Children with MBD5 deletion in particular have reduced glutathione production. Genetics likely plays a huge role in susceptibility to AR toxicity.
AR can’t be stored like retinol in hepatic stellate cells because it lacks a polar “-OH” group, and this feature also may make it unable to diffuse through cell membranes. However, it has been found to have a high affinity for RBP4 and CRBP. This would, in theory ,displace retinol from RBP4 and also CRBP leaving healthy retinol unbound. This cause a high primary alcohol state. PAPS is a buffer for primary alcohols, like ethanol, but there is no sulfotransferase available for retinol, and so retinol will remain free in the cytosol.
I think it’s probable that we have to get into a state of “low alcohol” in a cell to be able to use an acid to convert AR to rehydroretinol. I also think it’s possible that if there is unbound retinol in the cell, it can be the “alcohol” that is causing the problem of making more retinol. This means that having plenty of phosphatidylcholine available for LRAT to safely tuck away vitamin A is important. The back up enzymes ARAT and DGAT are also helpful, but I suspect these are tied up in fatty acid synthesis as AR pushes people into ketosis and also makes them produce excessive lipids from citrate exportation from the cells. It is also possible that the low vitamin A diet can help someone get into a low retinol state. However, my own person experience with my daughter over the past three years was that a low vitamin A diet did not solve her vitamin A toxicity, and in fact she worsened. Hind sight tells me that this could be due to the lack of unadulterated retinol to buffer anhydroretinol in the cells.
Cellular control of retinol levels is crucial to prevent cell toxicity. The ability to excrete retinol from cells onto RBP4 to keep cytosolic levels of retinol normal is highly dependent on blood pH. In acidosis, calcium channels function poorly and calcium influx into cells decreases. Calcium is crucial for the STRA6 pore that allows retinol to leave the cell to open. Calcium actually binds to calmodulin, and when calcium levels increase in the cell, STRA6 doors are open. Unfortunately, when AR takes the place of retinol in PKCδ and pathologically turns on PDHC, the ensuing reactive oxygen species pushes cells into reluctant ketosis which causes acidosis. Thus, efflux of retinol from cells is stagnant, and until acidosis is resolved, retinol levels will stay high in cells, or if the cell is in a low retinol state, the levels will stay low. It may be though, since retinol is polar, that it can diffuse out of the cell through membranes. This remains to be explored, but it is possible that while retinol is doing this, which isn’t very efficient at all.
What if most of the retinol in the body is AR or Rehydroretinol?
The possibility that someone is so very far stuck in this pathway that they have no true retinol is quite possible. What would this look like? An individual that lacks sufficient, true retinol would have immune dysfunction. Blood work would show IgA and/or IgG deficiency. Stool testing would show IgA deficiency. This is due to retinoic acid is needed for the immune system to function. A person with severe AR toxicity might have low lymphocyte counts and high liver enzymes. I suspect this person’s underlying issue would be candida of the stomach that hasn’t been diagnosed. This would lead to all oral intakes of vitamin A being converted into anhydroretinol while in the gut. Someone with mild AR toxicity might only have low platelet counts and high monocytes as retinoic acid is needed to induce platelet production and it is needed to convert monocytes into macrophages. Of course, these are general ideas based on experience and reading individual labs of the people I care for.
What made me find AR as the sneaky vitamin A toxin?
After an entire year dealing with clients suffering from vitamin A toxicity as well as my own daughter having suffered for three years, one of my clients, Oasis, who has a CAPZB variant showed me that the true problem is anhydroretinol. His mother and I have become good friends and we prayed that we would find what is causing Oasis, Zoey, and my other client’s inability to use vitamin A correctly in their body. Our prayers were answered.
Oasis’ CAPZB variant makes him have an altered versions of F-Actin which is a cytoskeletal protein that gives membranes stability. He is more vulnerable to anything that causes oxidative stress to membranes. He has suffered greatly over the past five years from anhydroretinol toxicity from a combination of enteral nutrition, valproic acid altering his ability to bind retinol in the body, and altered metabolism. We can all thank Oasis and his mom Marcela for helping us to solve the puzzle of vitamin A toxicity, but not just them!
I also need to thank a few other clients and parents for trusting me in this deep dive. Olivia’s labs (in addition to Oasis’) showed me all the worst broken pathways when vitamin A is out of control. My own daughter Zoey’s urinary organic acid tests and plasma amino acid tests were quite revealing. The trends were consistent among these individuals. And finally, James, who has down syndrome, also helped me to find what’s truly going on in Vitamin A toxicity. There are a few other clients that are no longer with me due to a change in my employment, but they also helped me along this journey to find that ALL ROADS LEAD TO ANHYDRORETINOL.
Of course, I can’t forget my excellent sounding board that consisted of Andrew Baird, vitamin A researcher, Jenny Jones, PhD, molecular genetics, Michelle Harris, nutrition student (close to graduation!), and Bryan Toh, pharmacist. Without them checking my hypotheses and feeding bits of information that I didn’t know, because, yes, we all can’t know everything, I wouldn’t have finally found the root cause of vitamin A toxicity.
What is the rescue?How do we escape from the devastating effects of AR?
(Please consult with your personal healthcare provider before making changes)
In short, okay, not so short, the rescue is:
Choline from phosphatidylcholine or eggs (I prefer free range eggs) – 500 mg of choline is the goal.
Meeting at minimum the RDA for minerals especially Cu, Mo, Zn, Se, Mn. (may need more or less)
Benfotiamine (down regulates NF-KB and lowers iNOS, restores pentose phosphate shunt generation of NADPH needed for glutathione recycling)
Pantothenic acid (avoidance of biotin or lipoate at the same time as pantothenic acid; helps to restore acetate to acetyl CoA instead of becoming acetone). I am unsure of the amount. I am giving my own daughter 25 mg per day.
Vitamin E (Possibly 400 IU per day. I don’t recommend going very high because we need enough vitamin C in the body to recycle the oxidized form of vitamin E.)
Vitamin C (preferably slow release vitamin C, 250 mg per day, or dietary sources of vitamin C)
Coenzyme Q10 (to mop up the excess electrons being produced in the cells from pathological turning on of PDHC)
B12 – 2000 mcg per day
Folate in the form of 5-methylfolate or folinic acid (although folinic acid may contribute to gout)
After making sure mineral are restored, increase betaine intake or start betaine (trimethylglycine) but only 250 mg for kids and 500 mg for adults if you have not had a plasma amino acid tests to check methionine levels. There is a small chance that people in this pathway have high slow CBS enzymes which can cause high methionine levels and adding betaine could cause brain edema. This is very rare, but any children that are non-speaking should have a plasma amino acid test prior to starting betaine and never go over 1000 mg per day.
At least 2 mg of riboflavin to support betaine pathway
A source of retinol daily. I believe that many people who are making too much anhydroretinol could just have rehydroretinol available, and this version of vitamin A has altered double bond configuration and probably will not function like retinol (researchers say 7% activity of retinol). One source of retinol could be the eggs – many don’t agree with me on this, but I have found that eggs have rescued my clients from high AST and ALT. Also, eggs are a good source of biotin and biotin is needed for the STRA6 pore to work properly. Taking a biotin supplement is problematic because it competes with pantothenic acid for absorption. Another possible source of retinol is raw milk or butter (be sure to follow food safety precautions and find a reputable source).
CAUTION with increase retinol in those with candida or other fungal infections. I think these people are the individuals who feel sick from consuming retinol sources. Treating fungal infections may be necessary before increasing retinol intake.
Treatment of candida (There is a risk for candida of the esophagus and stomach in anyone who takes antihistamines. This may a contributor to making AR in the intestinal track before absorption. This is what is happening to my daughter.)
Epsom salt baths to help PAPS (exploring PAPS due to APS pyruvate carboxylase)
Liposomal glutathione. This is controversial because it’s possible that the glutathione is in a oxidized form. However, oxidized glutathione actually triggers the CBS enzyme to upregulate and make more glutathione. Also, with the restoration of the pentose phosphate shunt pathway using benfotiamine, there should be plenty of NADPH to be able to restore glutathione to its reduced state.
Monitor for acidosis with health care practitioner and treat if able
AVOID citrate supplements or citrate versions of supplements
Preferable potassium bicarbonate or sodium bicarbonate (but not with meals)
acidosis needs to be resolved because “first pass” of fresh retinol that is needed by cells in the peripheral tissues is dependent on uptake of triglycerides from chylomicron first.
Things to avoid if you are susceptible to anhydroretinol toxicity…
Saccharomyces boulardii. This is an “anti-yeast” that people take when having candida. It is sometimes added to probiotics. This has been shown to make more alcohol than Candida
Alcohol – alcohol is the determinant factor in making anhydroretinol in the gut. Especially never eat cheese with your alcohol!
Spore based bacillus subtilis (metabolizes sulfate) and we actually need all the sulfate we can get to restore PAPS which takes care of excess alcohol in the cell. It won’t fix the high retinol problem. More on that in a later post.
Annatto and Saffron – contain apo-carotenal and this could be pathological if in PKCδ
Excess carotenoids if you KNOW your have a slow BCO1 because BCO2 makes various apo-carotenols which might displace retinol from PKCdelta.
One carotenol that is made from retinol, retinaldehyde, and retinoic acid, under oxidative stress is beta-apo-13′-carotenal that is known to inhibit CYP26 and also likely turns on PDCH pathologically. (doi: 10.3390/nu14071411;
“Shelf stable” foods with vitamin A added. Vitamin A degrades over time and the vitamin A added could have some anhydroretinol in it.
Skin products with retinyl palmitate or retinol added as the UV light makes anhydroretinol in skin.
Milk in clear containers. Avoid fat free milk that is fortified with vitamin A.
Cod liver oil. This was the first product in which scientists found anhydroretinol!
Any competitors for aldehyde oxidase. Aldehyde oxidase is the only enzyme to convert retinaldehyde to retinoic acid without requiring NAD. It is Mo dependent). Other substrates for AOX include niacin, caffeine, and vitamin B6. Often B6 levels will be high in the blood and that is a sneaky sign of having issues with anhydroretinol.
THE MASSIVE AMOUNT OF ARTICLES THAT I READ TO COME TO THIS CONCLUSION
Hammerling U, Kim YK, Quadro L. Quantum chemistry rules retinoid biology. Commun Biol. 2023 Feb 28;6(1):227. doi: 10.1038/s42003-023-04602-x. PMID: 36854887; PMCID: PMC9974979.
Hammerling U. Retinol as electron carrier in redox signaling, a new frontier in vitamin A research. Hepatobiliary Surg Nutr. 2016 Feb;5(1):15-28. doi: 10.3978/j.issn.2304-3881.2016.01.02. PMID: 26904553; PMCID: PMC4739943.
Buck J, Grün F, Derguini F, Chen Y, Kimura S, Noy N, Hämmerling U. Anhydroretinol: a naturally occurring inhibitor of lymphocyte physiology. J Exp Med. 1993 Aug 1;178(2):675-80. doi: 10.1084/jem.178.2.675. PMID: 8340762; PMCID: PMC2191109.
Acin-Perez R, Hoyos B, Zhao F, Vinogradov V, Fischman DA, Harris RA, Leitges M, Wongsiriroj N, Blaner WS, Manfredi G, Hammerling U. Control of oxidative phosphorylation by vitamin A illuminates a fundamental role in mitochondrial energy homoeostasis. FASEB J. 2010 Feb;24(2):627-36. doi: 10.1096/fj.09-142281. Epub 2009 Oct 7. PMID: 19812372; PMCID: PMC2812036.
Kim YK, Hammerling U. The mitochondrial PKCδ/retinol signal complex exerts real-time control on energy homeostasis. Biochim Biophys Acta Mol Cell Biol Lipids. 2020 Nov;1865(11):158614. doi: 10.1016/j.bbalip.2020.158614. Epub 2020 Jan 10. PMID: 31927141; PMCID: PMC7347429.
Korichneva I, Hämmerling U. F-actin as a functional target for retro-retinoids: a potential role in anhydroretinol-triggered cell death. J Cell Sci. 1999 Aug;112 ( Pt 15):2521-8. doi: 10.1242/jcs.112.15.2521. PMID: 10393808.
Md. Jakaria, Abdel A. Belaidi, Ashley I. Bush, Scott Ayton, Vitamin A metabolites inhibit ferroptosis [anhydroretinol sensitizes cells to ferroptosis], Biomedicine & PharOasistherapy, Volume 164, 2023, 114930, ISSN 0753-3322, https://doi.org/10.1016/j.biopha.2023.114930.
T.K. Murray, P. Erdody, The absorption and metabolism of anhydrovitamin A by the rat, Biochimica et Biophysica Acta (BBA) – General Subjects, Volume 136, Issue 2, 1967, Pages 375-378, ISSN 0304-4165, https://doi.org/10.1016/0304-4165(67)90082-7.
Martin Kohlmeier,Chapter 9 – Fat-Soluble Vitamins and Nonnutrients,Editor(s): Martin Kohlmeier,Nutrient Metabolism (Second Edition),Academic Press,2015,Pages479-565,ISBN 9780123877840,https://doi.org/10.1016/B978-0-12-387784-0.00009-2.
Gloria E. Mao, Michael D. Collins, Fadila Derguini, Teratogenicity, Tissue Distribution, and Metabolism of the retro-Retinoids, 14-Hydroxy-4,14-retro-retinol and Anhydroretinol, in the C57BL/6J Mouse,Toxicology and Applied PharOasislogy,Volume 163, Issue 1,2000,Pages 38-49,ISSN 0041-008X, https://doi.org/10.1006/taap.1999.8828.
O’Connell MJ, Chua R, Hoyos B, Buck J, Chen Y, Derguini F, Hämmerling U. Retro-retinoids in regulated cell growth and death. J Exp Med. 1996 Aug 1;184(2):549-55. doi: 10.1084/jem.184.2.549. PMID: 8760808; PMCID: PMC2192720.
Chen Y, Buck J, Derguini F. Anhydroretinol induces oxidative stress and cell death. Cancer Res. 1999 Aug 15;59(16):3985-90. PMID: 10463596.
Chiu HJ, Fischman DA, Hammerling U. Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP-1-dependent energy deprivation. FASEB J. 2008 Nov;22(11):3878-87. doi: 10.1096/fj.08-112375. Epub 2008 Aug 1. PMID: 18676402; PMCID: PMC2574026.
de Oliveira MR. Vitamin A and Retinoids as Mitochondrial Toxicants. Oxid Med Cell Longev. 2015;2015:140267. doi: 10.1155/2015/140267. Epub 2015 May 19. PMID: 26078802; PMCID: PMC4452429.
Klamt F, Dal-Pizzol F, Gelain DP, Dalmolin RS, Birnfeld de Oliveira R, Bastiani M, Horn F, Fonseca Moreira JC. Vitamin A treatment induces apoptosis through an oxidant-dependent activation of the mitochondrial pathway. Cell Biol Int. 2008 Jan;32(1):100-6. doi: 10.1016/j.cellbi.2007.08.018. Epub 2007 Sep 7. PMID: 17942326.
Álvarez, R., Vaz, B., Gronemeyer, H., & de Lera, A.R. (2014). Functions, therapeutic applications, and synthesis of retinoids and carotenoids. Chemical reviews, 114 1, 1-125 .
Yokoyama H, Matsumoto M, Shiraishi H, Miyagi M, Kato And S, Ishii H. Nicotinamide adenine dinucleotide-dependent retinoic acid formation from retinol in the human gastric mucosa: inhibition by ethanol, acetaldehyde, and H2 blockers. Alcohol Clin Exp Res. 2001 Jun;25(6 Suppl):24S-8S. doi: 10.1097/00000374-200106001-00007. PMID: 11410737.
Ramkumar S, Moon J, Golczak M, von Lintig J. LRAT coordinates the negative-feedback regulation of intestinal retinoid biosynthesis from β-carotene. J Lipid Res. 2021;62:100055. doi: 10.1016/j.jlr.2021.100055. Epub 2021 Feb 23. PMID: 33631212; PMCID: PMC8010212.
Bhat PV, Roller PP, De Luca LM. Chemical and biological studies on 5,6-epoxyretinol, retinol, and their phosphoryl esters. J Lipid Res. 1981 Sep;22(7):1069-78. PMID: 7299288.
Zweier, Jay & Velayutham, Murugesan & Hemann, Craig. (2013). FRBM4.
Tejada-Jimenez M, Chamizo-Ampudia A, Calatrava V, Galvan A, Fernandez E, Llamas A. From the Eukaryotic Molybdenum Cofactor Biosynthesis to the Moonlighting Enzyme mARC. Molecules. 2018; 23(12):3287. https://doi.org/10.3390/molecules23123287
Wang CH, Zhang C, Xing XH. Xanthine dehydrogenase: An old enzyme with new knowledge and prospects. Bioengineered. 2016 Nov;7(6):395-405. doi: 10.1080/21655979.2016.1206168. Epub 2016 Aug 18. PMID: 27537049; PMCID: PMC5094624.
Jansson EA, Huang L, Malkey R, Govoni M, Nihlén C, Olsson A, Stensdotter M, Petersson J, Holm L, Weitzberg E, Lundberg JO. A mammalian functional nitrate reductase that regulates nitrite and nitric oxide homeostasis. Nat Chem Biol. 2008 Jul;4(7):411-7. doi: 10.1038/nchembio.92. Epub 2008 May 30. PMID: 18516050.
Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997 Sep 26;272(39):24154-8. doi: 10.1074/jbc.272.39.24154. PMID: 9305864.
Zeczycki TN, Maurice MS, Attwood PV. Inhibitors of Pyruvate Carboxylase. Open Enzym Inhib J. 2010;3:8-26. doi: 10.2174/1874940201003010008. PMID: 22180764; PMCID: PMC3238542.
Moffett JR, Puthillathu N, Vengilote R, Jaworski DM, Namboodiri AM. Acetate Revisited: A Key Biomolecule at the Nexus of Metabolism, Epigenetics and Oncogenesis-Part 1: Acetyl-CoA, Acetogenesis and Acyl-CoA Short-Chain Synthetases. Front Physiol. 2020 Nov 12;11:580167. doi: 10.3389/fphys.2020.580167. PMID: 33281616; PMCID: PMC7689297.
Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014 Jul;71(14):2577-604. doi: 10.1007/s00018-013-1539-2. Epub 2013 Dec 21. PMID: 24363178; PMCID: PMC4059968.
Velayutham M, Hemann CF, Cardounel AJ, Zweier JL. Sulfite Oxidase Activity of Cytochrome c: Role of Hydrogen Peroxide. Biochem Biophys Rep. 2016 Mar 1;5:96-104. doi: 10.1016/j.bbrep.2015.11.025. PMID: 26709389; PMCID: PMC4689149.
Kundu TK, Velayutham M, Zweier JL. Aldehyde oxidase functions as a superoxide generating NADH oxidase: an important redox regulated pathway of cellular oxygen radical formation. Biochemistry. 2012 Apr 3;51(13):2930-9. doi: 10.1021/bi3000879. Epub 2012 Mar 19. PMID: 22404107; PMCID: PMC3954720.
Kitamura S, Sugihara K, Ohta S. Drug-metabolizing ability of molybdenum hydroxylases. Drug Metab Pharmacokinet. 2006 Apr;21(2):83-98. doi: 10.2133/dmpk.21.83. PMID: 16702728.
Appendix C: Dissociation Constants and pKa Values for Acids at 25°C”, appendix 3 from the book Principles of General Chemistry (v. 1.0).
Tolleson WH, Cherng SH, Xia Q, Boudreau M, Yin JJ, Wamer WG, Howard PC, Yu H, Fu PP. Photodecomposition and phototoxicity of natural retinoids. Int J Environ Res Public Health. 2005 Apr;2(1):147-55. doi: 10.3390/ijerph2005010147. PMID: 16705812; PMCID: PMC3814709.
Álvarez, R., Vaz, B., Gronemeyer, H., & de Lera, A.R. (2014). Functions, therapeutic applications, and synthesis of retinoids and carotenoids. Chemical reviews, 114 1, 1-125 .
Kinetics of Formation of Anhydroretinol From Retinyl Acetate in Acetic Acid, and From Retinyl Acetate and Retinol in Ethanol and Ethanol–Water Mixtures Containing Hydrogen Chloride.Acta Chemica Scandinavicadoi 10.3891/acta.chem.scand.30a-0285
Saeed A, Bartuzi P, Heegsma J, Dekker D, Kloosterhuis N, de Bruin A, Jonker JW, van de Sluis B, Faber KN. Impaired Hepatic Vitamin A Metabolism in NAFLD Mice Leading to Vitamin A Accumulation in Hepatocytes. Cell Mol Gastroenterol Hepatol. 2021;11(1):309-325.e3. doi: 10.1016/j.jcmgh.2020.07.006. Epub 2020 Jul 19. PMID: 32698042; PMCID: PMC7768561.
Ciorba MA. Kynurenine pathway metabolites: relevant to vitamin B-6 deficiency and beyond. Am J Clin Nutr. 2013 Oct;98(4):863-4. doi: 10.3945/ajcn.113.072025. Epub 2013 Aug 28. PMID: 23985806; PMCID: PMC4498264.
Sherriff JL, O’Sullivan TA, Properzi C, Oddo JL, Adams LA. Choline, Its Potential Role in Nonalcoholic Fatty Liver Disease, and the Case for Human and Bacterial Genes. Adv Nutr. 2016 Jan 15;7(1):5-13. doi: 10.3945/an.114.007955. PMID: 26773011; PMCID: PMC4717871.
Harrison EH, Quadro L. Apocarotenoids: Emerging Roles in Mammals. Annu Rev Nutr. 2018 Aug 21;38:153-172. doi: 10.1146/annurev-nutr-082117-051841. Epub 2018 May 11. PMID: 29751734; PMCID: PMC6295211.
Fortuna VA, Trugo LC, Borojevic R. Acyl-CoA: retinol acyltransferase (ARAT) and lecithin:retinol acyltransferase (LRAT) activation during the lipocyte phenotype induction in hepatic stellate cells. J Nutr Biochem. 2001 Nov;12(11):610-621. doi: 10.1016/s0955-2863(01)00179-6. PMID: 12031254.
Shih MY, Kane MA, Zhou P, Yen CL, Streeper RS, Napoli JL, Farese RV Jr. Retinol Esterification by DGAT1 Is Essential for Retinoid Homeostasis in Murine Skin. J Biol Chem. 2009 Feb 13;284(7):4292-9. doi: 10.1074/jbc.M807503200. Epub 2008 Nov 20. PMID: 19028692; PMCID: PMC2640966.
Lillig, Christopher Horst & Fernandes, Aristi & Schwenn, Jens & Vlamis-Gardikas, Alexios & Holmgren, Arne. (2003). Redox Regulation of 3′-Phosphoadenylylsulfate Reductase from Escherichia coli by Glutathione and Glutaredoxins. The Journal of biological chemistry. 278. 22325-30. 10.1074/jbc.M302304200.
Carnauba RA, Baptistella AB, Paschoal V, Hübscher GH. Diet-Induced Low-Grade Metabolic Acidosis and Clinical Outcomes: A Review. Nutrients. 2017 May 25;9(6):538. doi: 10.3390/nu9060538. PMID: 28587067; PMCID: PMC5490517.
Yamada K, Yoshida K. Multiple subcellular localizations and functions of protein kinase Cδ in liver cancer. World J Gastroenterol. 2022 Jan 14;28(2):188-198. doi: 10.3748/wjg.v28.i2.188. PMID: 35110944; PMCID: PMC8776529.
Boer, Johannes & Alsters, Paul & Meetsma, Auke & Hage, Ronald & Browne, Wesley & Feringa, Ben. (2008). The role of salicylic acid, L-ascorbic acid and oxalic acid in promoting the oxidation of alkenes with H2O2 catalysed by [MnIV2(O)3(tmtacn)2]2+. Dalton transactions (Cambridge, England : 2003). 44. 6283-95. 10.1039/b809177c.
Webster CR, Johnston AN, Anwer MS. Protein kinase Cδ protects against bile acid apoptosis by suppressing proapoptotic JNK and BIM pathways in human and rat hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2014 Dec 15;307(12):G1207-15. doi: 10.1152/ajpgi.00165.2014. Epub 2014 Oct 30. PMID: 25359536; PMCID: PMC4269680.
Leitges, Michael & Gimborn, Kerstin & Elis, Winfried & Kalesnikoff, Janet & Hughes, Michael & Krystal, Gerald & Huber, Michael. (2002). Protein Kinase C- Is a Negative Regulator of Antigen-Induced Mast Cell Degranulation. Molecular and cellular biology. 22. 3970-80. 10.1128/MCB.22.12.3970-3980.2002.
Penniston, Kristina & Tanumihardjo, Sherry. (2006). The acute and chronic effects of vitamin A. The American journal of clinical nutrition. 83. 191-201. 10.1093/ajcn/83.2.191.
O’Connell MJ, Chua R, Hoyos B, Buck J, Chen Y, Derguini F, Hämmerling U. Retro-retinoids in regulated cell growth and death. J Exp Med. 1996 Aug 1;184(2):549-55. doi: 10.1084/jem.184.2.549. PMID: 8760808; PMCID: PMC2192720.
Gimeno A, Zaragozá R, Vivó-Sesé I, Viña JR, Miralles VJ. Retinol, at concentrations greater than the physiological limit, induces oxidative stress and apoptosis in human dermal fibroblasts. Exp Dermatol. 2004 Jan;13(1):45-54. doi: 10.1111/j.0906-6705.2004.00112.x. PMID: 15009115.
Venkatachalam, Kallidaikurichi. (2015). Biochemical Sulfuryl Group Transfer From 3’-Phosphoadenosine 5’-Phosphosulfate (PAPS) Versus Phosphoryl Transfer From ATP: What Can Be Learnt?. Biochemistry and Physiology. 5. 10.4172/2168-9652.1000192.
MAHTAB S. BAMJI, H. R. CAMA, AND P. R. SUNDARESAN. THE JOURNAL CIBOLO(ICAL CHEMISTRY Anhydrovitamin A, and Rehydrovitamin A,. II Vol. 237, No. 9,September 1962 Printed in U.S.A.
P. Phuapradit, M.R. Lakshmanan, J.A. Olson,The excretion of polar metabolites of radioactive anhydroretinol in rabbit bile,Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism,Volume 260, Issue 4,1972,Pages 666-669,ISSN 0005-2760, https://doi.org/10.1016/0005-2760(72)90015-X.
Philip D. Kiser, Marcin Golczak, and Krzysztof Palczewski Chemistry of the Retinoid (Visual) Cycle Chemical Reviews 2014 114 (1), 194-232 DOI: 10.1021/cr400107q
Minren Xu and Jim WatsonMicroencapsulated Vitamin A Palmitate Degradation Mechanism Study To Improve the Product Stability Journal of Agricultural and Food Chemistry 2021 69 (51), 15681-15690 DOI: 10.1021/acs.jafc.1c06087
Yang H, Xu L, Hou L, Xu TC, Ye SH. Stability of vitamin A, E, C and thiamine during storage of different powdered enteral formulas. Heliyon. 2022 Nov 12;8(11):e11460. doi: 10.1016/j.heliyon.2022.e11460. PMID: 36411896; PMCID: PMC9674494.
Mao GE, Collins MD, Derguini F. Teratogenicity, tissue distribution, and metabolism of the retro-retinoids, 14-hydroxy-4,14-retro-retinol and anhydroretinol, in the C57BL/6J mouse. Toxicol Appl PharOasisl. 2000 Feb 15;163(1):38-49. doi: 10.1006/taap.1999.8828. PMID: 10662603.
Zhong G, Seaman CJ, Paragas EM, Xi H, Herpoldt KL, King NP, Jones JP, Isoherranen N. Aldehyde Oxidase Contributes to All-Trans-Retinoic Acid Biosynthesis in Human Liver. Drug Metab Dispos. 2021 Mar;49(3):202-211. doi: 10.1124/dmd.120.000296. Epub 2020 Dec 18. PMID: 33355213; PMCID: PMC7885020.
Modin A, Björne H, Herulf M, Alving K, Weitzberg E, Lundberg JO. Nitrite-derived nitric oxide: a possible mediator of ‘acidic-metabolic’ vasodilation. Acta Physiol Scand. 2001 Jan;171(1):9-16. doi: 10.1046/j.1365-201X.2001.00771.x. PMID: 11350258.
Flores-Cortez YA, Barragán-Bonilla MI, Mendoza-Bello JM, González-Calixto C, Flores-Alfaro E, Espinoza-Rojo M. Interplay of retinol binding protein 4 with obesity and associated chronic alterations (Review). Mol Med Rep. 2022 Jul;26(1):244. doi: 10.3892/mmr.2022.12760. Epub 2022 Jun 3. PMID: 35656886; PMCID: PMC9185696.
Bozic I, Savic D, Laketa D, Bjelobaba I, Milenkovic I, Pekovic S, Nedeljkovic N, Lavrnja I. Benfotiamine attenuates inflammatory response in LPS stimulated BV-2 microglia. PLoS One. 2015 Feb 19;10(2):e0118372. doi: 10.1371/journal.pone.0118372. PMID: 25695433; PMCID: PMC4335016.
Veselá A, Wilhelm J. The role of carbon dioxide in free radical reactions of the organism. Physiol Res. 2002;51(4):335-9. PMID: 12449430.
Ciorba MA. Kynurenine pathway metabolites: relevant to vitamin B-6 deficiency and beyond. Am J Clin Nutr. 2013 Oct;98(4):863-4. doi: 10.3945/ajcn.113.072025. Epub 2013 Aug 28. PMID: 23985806; PMCID: PMC4498264.
Robert Harker DM, Martinez B, Tabaac BJ. B12 Deficiency and Clinical Presentation in the Setting of Nitric Oxide Use. Case Rep Neurol Med. 2021 Apr 8;2021:5590948. doi: 10.1155/2021/5590948. PMID: 33927908; PMCID: PMC8049814.
Amiraslani B, Sabouni F, Abbasi S, Nazem H, Sabet M. Recognition of betaine as an inhibitor of lipopolysaccharide-induced nitric oxide production in activated microglial cells. Iran Biomed J. 2012;16(2):84-9. doi: 10.6091/ibj.1012.2012. PMID: 22801281; PMCID: PMC3600952.
Nicolaou A, Kenyon SH, Gibbons JM, Ast T, Gibbons WA. In vitro inactivation of mammalian methionine synthase by nitric oxide. Eur J Clin Invest. 1996 Feb;26(2):167-70. doi: 10.1046/j.1365-2362.1996.122254.x. PMID: 8904527.
Sharma VS, Pilz RB, Boss GR, Magde D. Reactions of nitric oxide with vitamin B12 and its precursor, cobinamide. Biochemistry. 2003 Jul 29;42(29):8900-8. doi: 10.1021/bi034469t. PMID: 12873151.
Broderick KE, Singh V, Zhuang S, Kambo A, Chen JC, Sharma VS, Pilz RB, Boss GR. Nitric oxide scavenging by the cobalamin precursor cobinamide. J Biol Chem. 2005 Mar 11;280(10):8678-85. doi: 10.1074/jbc.M410498200. Epub 2005 Jan 4. PMID: 15632180.
Danishpajooh IO, Gudi T, Chen Y, Kharitonov VG, Sharma VS, Boss GR. Nitric oxide inhibits methionine synthase activity in vivo and disrupts carbon flow through the folate pathway. J Biol Chem. 2001 Jul 20;276(29):27296-303. doi: 10.1074/jbc.M104043200. Epub 2001 May 22. PMID: 11371572.
Sampaio AL, Dalli J, Brancaleone V, D’Acquisto F, Perretti M, Wheatley C. Biphasic modulation of NOS expression, protein and nitrite products by hydroxocobalamin underlies its protective effect in endotoxemic shock: downstream regulation of COX-2, IL-1β, TNF-α, IL-6, and HMGB1 expression. Mediators Inflamm. 2013;2013:741804. doi: 10.1155/2013/741804. Epub 2013 May 28. PMID: 23781123; PMCID: PMC3679756.
Weinberg JB, Chen Y, Jiang N, Beasley BE, Salerno JC, Ghosh DK. Inhibition of nitric oxide synthase by cobalamins and cobinamides. Free Radic Biol Med. 2009 Jun 15;46(12):1626-32. doi: 10.1016/j.freeradbiomed.2009.03.017. Epub 2009 Mar 27. Erratum in: Free Radic Biol Med. 2011 Oct 1;51(7):1471. PMID: 19328848; PMCID: PMC2745708.
Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Olin-Sandoval V, Grüning NM, Krüger A, Tauqeer Alam M, Keller MA, Breitenbach M, Brindle KM, Rabinowitz JD, Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015 Aug;90(3):927-63. doi: 10.1111/brv.12140. Epub 2014 Sep 22. PMID: 25243985; PMCID: PMC4470864.
Young BD, Varney KM, Wilder PT, Costabile BK, Pozharski E, Cook ME, Godoy-Ruiz R, Clarke OB, Mancia F, Weber DJ. Physiologically Relevant Free Ca2+ Ion Concentrations Regulate STRA6-Calmodulin Complex Formation via the BP2 Region of STRA6. J Mol Biol. 2021 Nov 5;433(22):167272. doi: 10.1016/j.jmb.2021.167272. Epub 2021 Sep 27. PMID: 34592217; PMCID: PMC8568335.
O’Connor C, Varshosaz P, Moise AR. Mechanisms of Feedback Regulation of Vitamin A Metabolism. Nutrients. 2022 Mar 21;14(6):1312. doi: 10.3390/nu14061312. PMID: 35334970; PMCID: PMC8950952.
Harrison EH. Carotenoids, β-Apocarotenoids, and Retinoids: The Long and the Short of It. Nutrients. 2022 Mar 28;14(7):1411. doi: 10.3390/nu14071411. PMID: 35406024; PMCID: PMC9003029.
Durojaye BO, Riedl KM, Curley RW Jr, Harrison EH. Uptake and metabolism of β-apo-8′-carotenal, β-apo-10′-carotenal, and β-apo-13-carotenone in Caco-2 cells. J Lipid Res. 2019 Jun;60(6):1121-1135. doi: 10.1194/jlr.M093161. Epub 2019 Mar 6. PMID: 30846527; PMCID: PMC6547639.