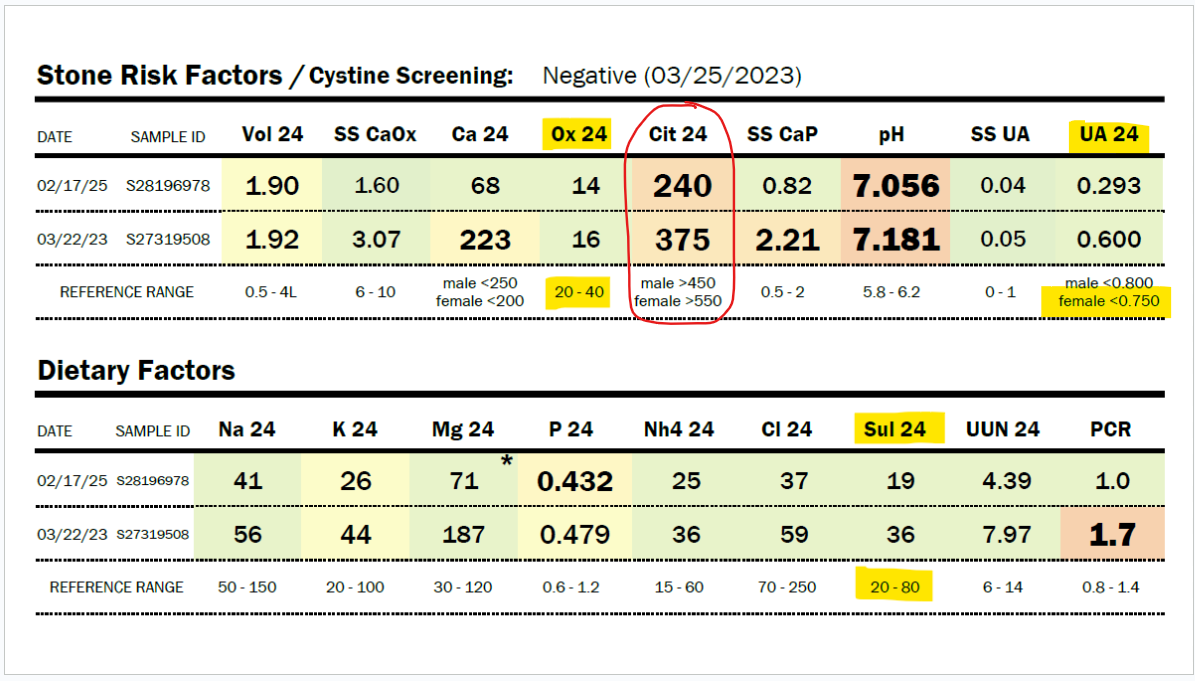
What if your sulfur metabolism issues aren’t due to sulfite oxidase or molybdenum cofactor deficiency? Read on to learn about how there may be a different cause to your sulfur intolerance!
***This is not medical advice but written for informational purposes only. Please consult with your personal healthcare provider prior to making any changes to your diet, supplements, medications, or lifestyle.***
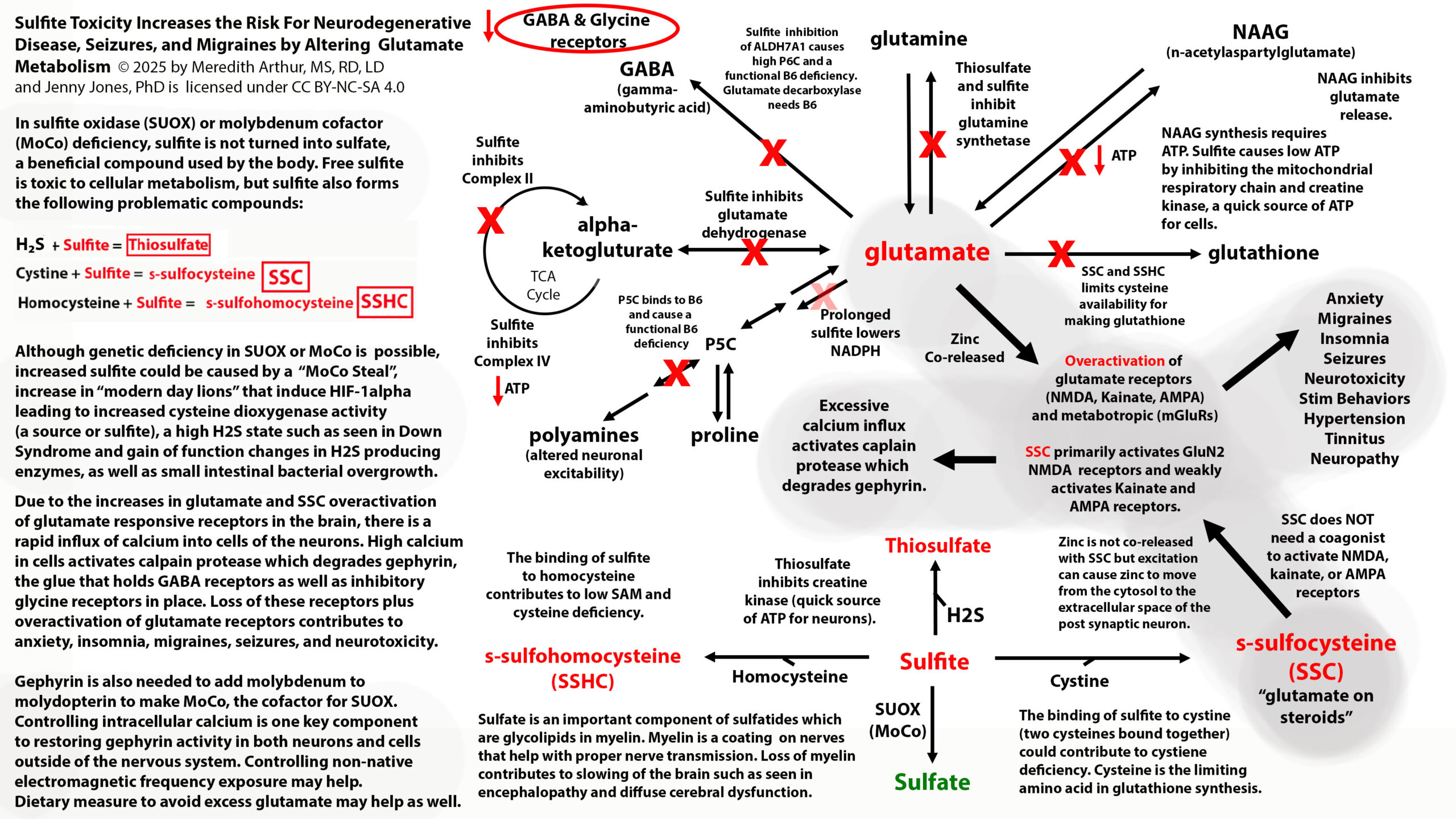
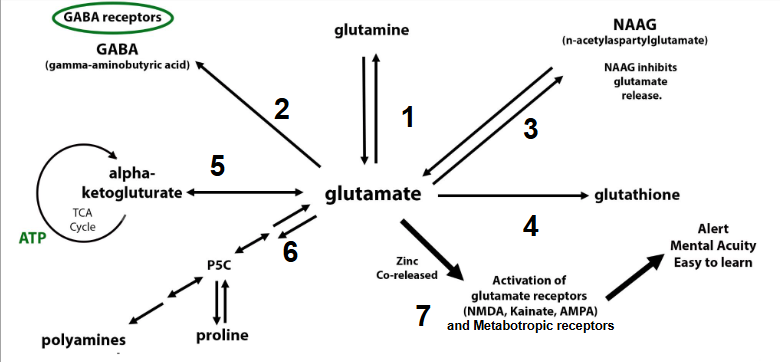
In November of 2024, the MoCo Steal Theory: Recovering From Sulfite Toxicity Support Group, was started by myself, Meredith Arthur, MS, RD, LD, and Jenny Jones, PhD. Jenny’s theory that due to high dose vitamin A intake, molybdenum cofactor could be stolen away from sulfite oxidase leading to sulfite toxicity, explained why she developed late-onset seizure disorder after using a vitamin A supplement. Her theory is that vitamin A in excess can shift molybdenum cofactor (MoCo) away from sulfite oxidase, and towards the backup enzyme for vitamin A metabolism in the liver, aldehyde oxidase. This would lead to higher levels of sulfite, which can cause a functional B6 deficiency by inhibiting ALDH7A1. This would increase glutamate levels in the brain, causing a seizure similar to pyridoxal-dependent epilepsy in those with classic ALDH7A1 deficiency. In addition this could lead to more s-sulfocysteins (SSC) which is like glutamate on steroids and can overexcite NMDA receptors in the brain.
However, she did not immediately develop seizure disorder from the vitamin A supplement, but this occurred after trying a low vitamin A, low choline diet to “detox” the vitamin A from her liver. Since starting this group, we have found that it’s not only the MoCo Steal that can contribute to sulfite toxicity by stealing MoCo from SUOX, causing intracellular sulfite toxicity and reaction to all things sulfur-related, but that there are also individuals who have extracellular sulfite toxicity as their driving force behind “sulfur intolerance”. We believe that going on a low vitamin A diet, or struggling with vitamin A metabolism in the gut, can shift the gut-associated lymphoid tissue away from an adaptive immune response into a neutrophil-dominated immune response that leads to extracellular sulfite toxicity.
INTRACELLULAR SULFITE TOXICITY
SUOX/MoCo deficiency.
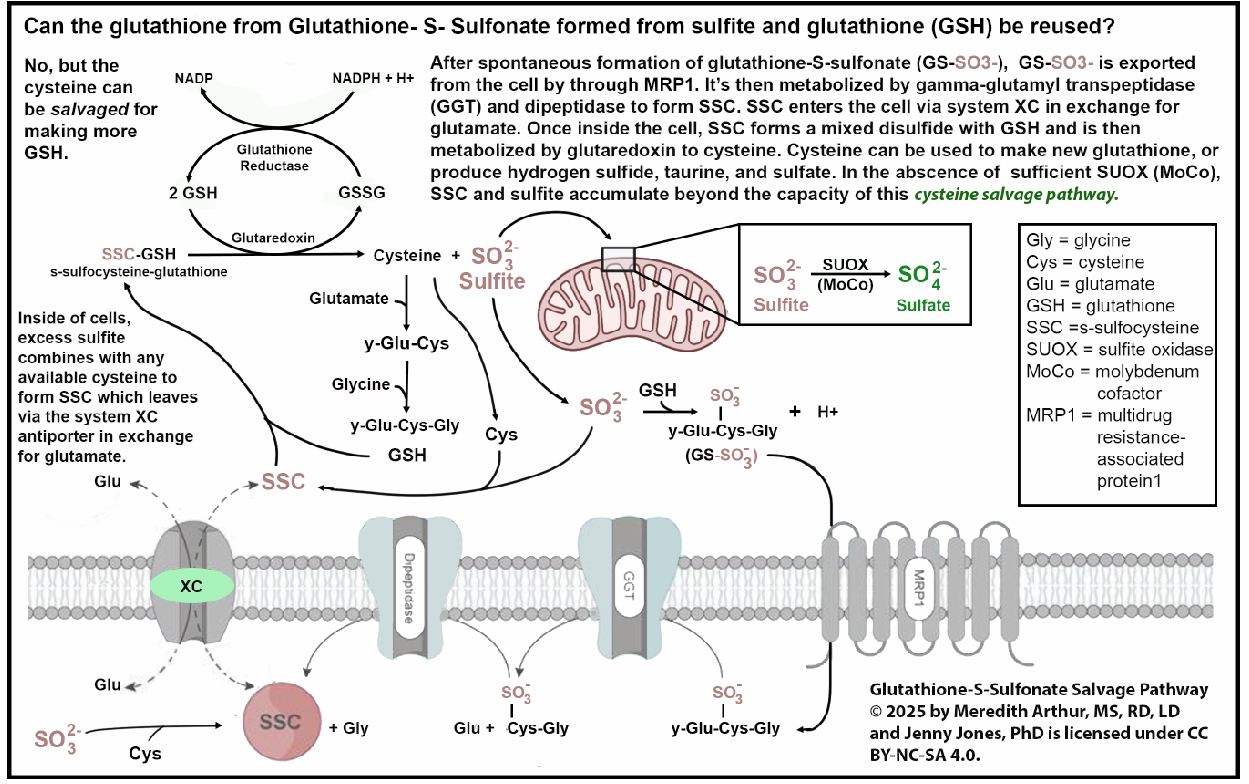
Some of us in the MoCo Steal: recovering from sulfite toxicity group on Facebook (https://www.facebook.com/share/g/17NqPUMKZj/) are struggling with sulfite due to burnout or inadequate SUOX function (a MoCo-dependent enzyme). Meaning that the amount of sulfite being produced by the transsulfuration pathway is exceeding our ability to convert it to sulfate, or because of increased activity of CDO due to upregulation of the HIF-1alpha pathway, we have excessive sulfite production by CDO. This causes high cytosolic and mitochondrial sulfite levels. This can lead to very high levels of s-sulfocysteine, SSC. We react to sulfur foods, garlic, NAC, and glutathione supplements.
EXTRACELLULAR SULFITE TOXICITY
Neutrophilic Sulfite Toxicity.
Some of the individuals in the MoCo Steal: Recovering from sulfite toxicity group on Facebook have lost their adaptive immune system (many have done the low vitamin A diet, which was a major contributing factor to loss of secretory IgA production and adaptive immune function). This causes the innate immune system (neutrophils and macrophages) to become dominant. In addition, many in this group struggle with SIBO, which provides additional H2S for neutrophils to use for sulfite production. In the portal vein, this excess sulfite can destroy B1 and B6 before they reach the liver and general circulation.
These individuals don’t have high levels of SSC because they don’t have SUOX deficiency or intracellular sulfite toxicity. The sulfite itself cannot cross cell membranes by diffusion. However, sulfite can increase the activity of NADPH oxidase in M1 polarized macrophages and increase macrophage phagocytosis. NADPH oxidase increases the production of superoxide.
Superoxide can’t diffuse across cell membranes into other cells, but when it’s metabolized to hydrogen peroxide via superoxide dismutase (SOD3) or when it spontaneously dismutates to hydrogen peroxide, this hydrogen peroxide can cross membranes. When superoxide builds up in macrophages, peroxynitrite (ONOO) increases. ONOO can also diffuse across phospholipid bilayers of cell membranes. ONOO and hydrogen peroxide are detoxified by glutathione peroxidase. This could lead to cellular glutathione deficiency, however if hydroxyl radicals are high, glutathione peroxidase activity will be inhibited.
Excessive neutrophil activation can lead to high levels of sulfate and sulfite radicals as well as hypochlorous acid. All of these metabolites can cause plasma glutathione deficiency because sulfite radicals and sulfate radicals irreversibly bind to glutathione in the blood.
We react to sulfur foods, garlic, NAC, and glutathione supplements because they provide ingredients that can increase sulfite production from neutrophils.
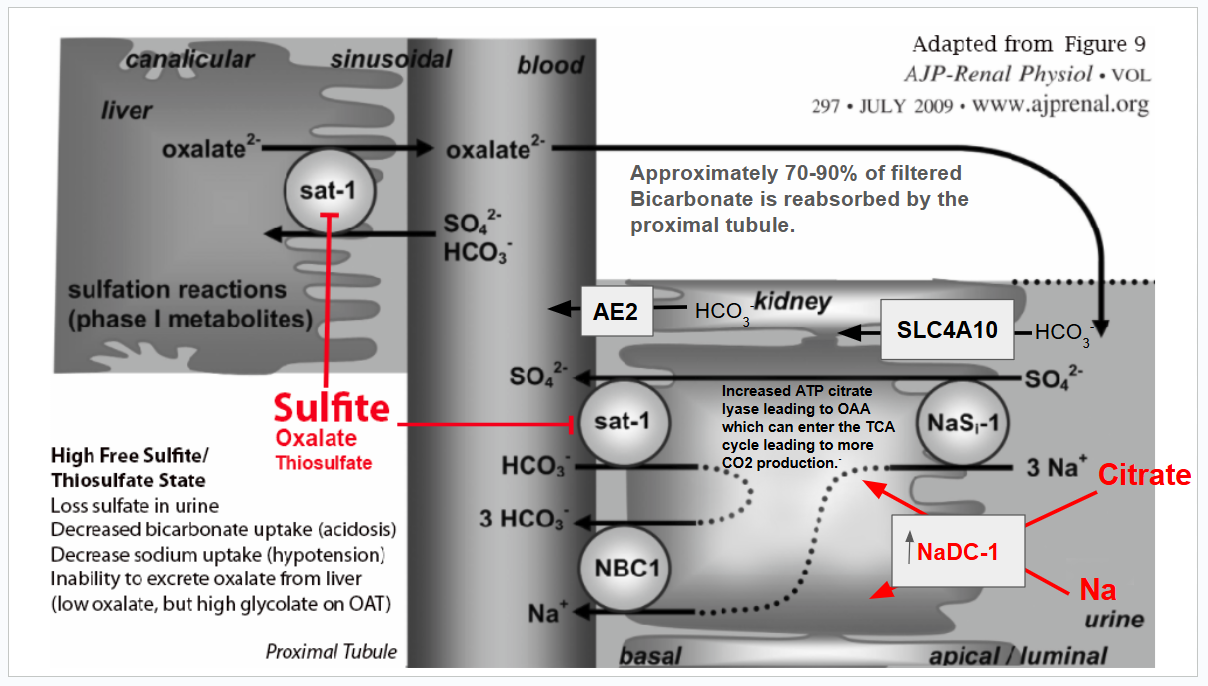
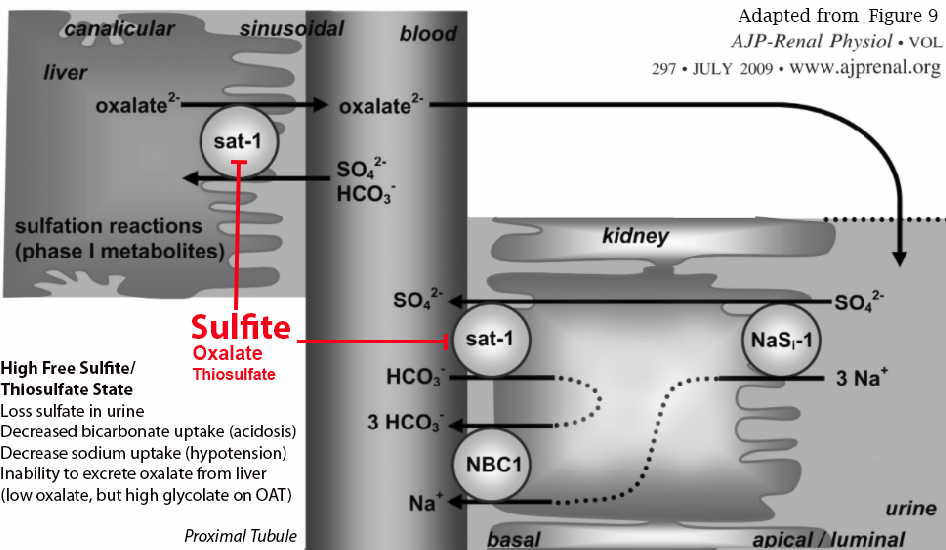
Below is a diagram showing the neutrophilic contribution of sulfite during the immune response.
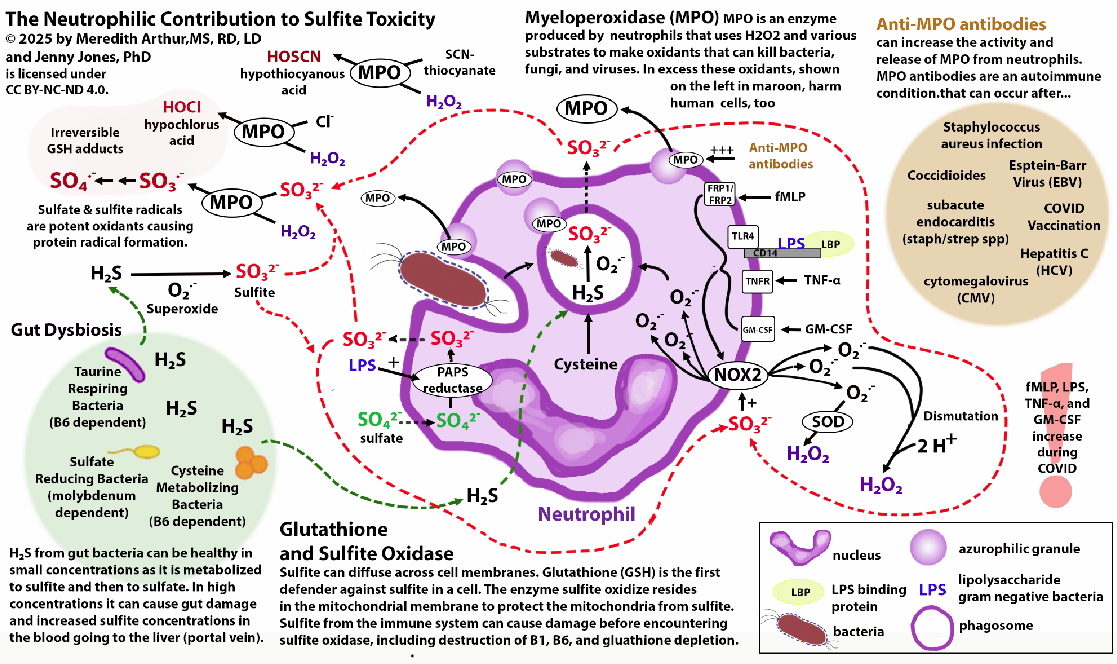
Neutrophils produce sulfite in response to LPS or when dealing with bacteria.
https://pubmed.ncbi.nlm.nih.gov/9823763
https://pubmed.ncbi.nlm.nih.gov/12512997
https://pubmed.ncbi.nlm.nih.gov/16317383
The overachiever. Some people are doing both! They have lost their adaptive immune system due to malnutrition as well and now rely on neutrophils and macrophages to deal with viruses and bacteria. Often, these people have very high sulfite production from neutrophils, leading to vitamin B1 deficiency. This causes intracellular B1 deficiency, leading to increased pyruvate levels, which includes the HIF-1alpha pathway and upregulation of CDO, leading to intracellular sulfite toxicity in the setting of overall inadequate nutrient intakes to be able to support molybdenum cofactor and sulfite oxidase enzyme production.
Why is it important to find out which group you are in?
If you are not high in SSC, then whatever diet you are currently on is specifically adequate in molybdenum. It means you are likely converting excess sulfite to sulfate in cells. It means that your cysteine salvage pathway, where you can salvage cysteine from SSC and then metabolize sulfite to sulfate, is working as well.
In addition, increasing molybdenum could support hydrogen sulfide-producing bacteria in the small intestine (small intestinal bacteria overgrowth with desulfovibrio species or bilophila). The H2S can fuel neutrophil production of sulfite, which will worsen the overall extracellular sulfite toxicity.
If you have normal SSC, you likely don’t need to supplement with molybdenum beyond what you are currently doing, and, for some, you may be able to back off on high doses (TALK WITH YOUR PRACTITIONER) as long-term high exposure to molybdenum is found to cause chronic kidney disease.
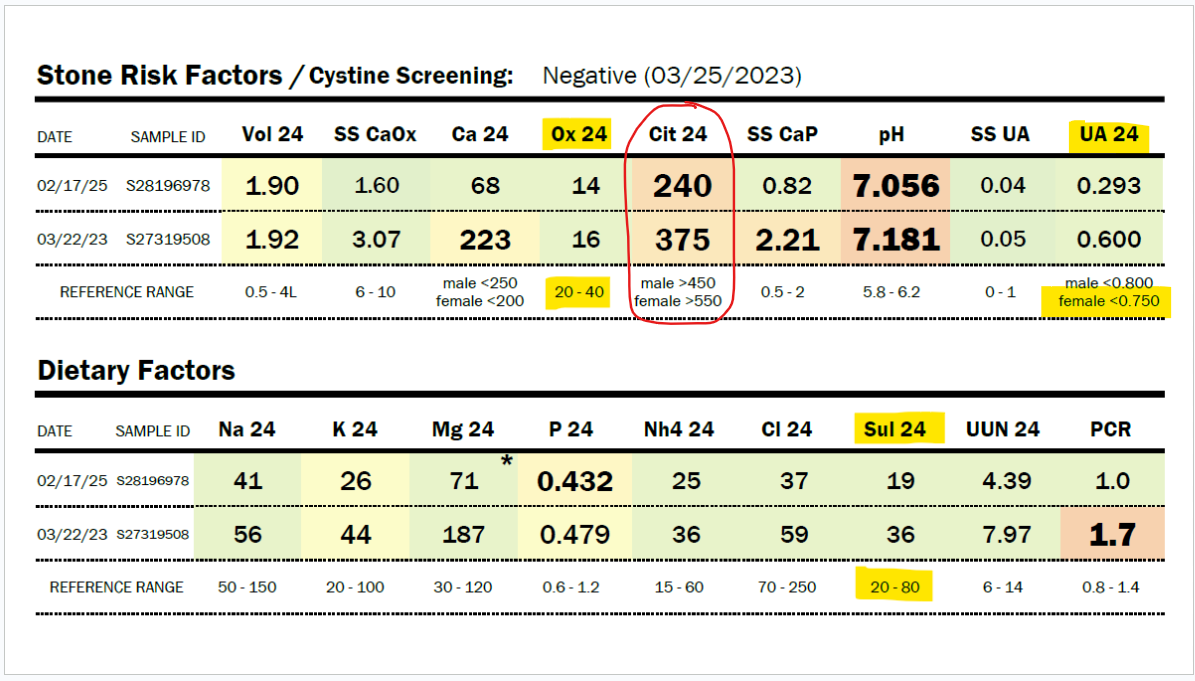
SSC can be tested via OMX from Diagnostic Solutions Laboratory.
https://www.diagnosticsolutionslab.com/tests/omx
How does excess molybdenum cause damage to the kidneys?
Molybdenum induces oxidative stress in renal tubular cells. The oxidative stress upregulates pro-apoptotic genes (cell death genes) and decreases anti-apoptotic genes (cell saving genes).
https://pubmed.ncbi.nlm.nih.gov/33348253
https://pubmed.ncbi.nlm.nih.gov/31811574
https://pubmed.ncbi.nlm.nih.gov/31518807
https://pubmed.ncbi.nlm.nih.gov/25627418
Molybdenum also promotes inflammatory responses in the kidney through the JAK/STAT axis and pyroptosis (swelling, explosion, and release of inflammatory contents) via the NLRP3/caspase-1 pathway.
https://pubmed.ncbi.nlm.nih.gov/33157513
https://pubmed.ncbi.nlm.nih.gov/38104808
https://pubmed.ncbi.nlm.nih.gov/34479002
Below is a slide of a summary slide of “Making MoCo” which is the cofactor needed for sulfite oxidase (SUOX). I made the “ingredients” in green. Promise I am making the video to explain how making MoCo can go wrong, but definitely working on the nutrients needed is very helpful. The goal is to absorb MoCo (see this video…. https://youtu.be/tA5ImU2vJKw) and then incorporated into MoCo where it should be!
Something I have found helpful to prevent excess H2S from bacteria binding to my molybdenum (makes tetrathiomolybdate) is to either take it on an empty stomach away from meals or take it with an apple which should in theory decrease H2S production (Chris Masterjohn talks about this in his sulfur protocol).
Ingredients for MoCo (listed in case English isn’t your native language and the slide is hard to read or if you have low vision and needed your phone to read the post):
- molybdenum (molybdate)
- radical SAMe (iron-sulfur cluster dependent)
- iron-sulfurs clusters (need B6 for formation)
- iron (need for heme component of SUOX, needed for iron sulfur cluster formation)
- B12, folate, and betaine (needed for methionine salvage pathway OR just adequate methionine intake)
- Magnesium (MPT synthase, and adding molybdenum via gephyrin)
- Zinc (stabilizes MPT synthase)
- Copper (tiny bit for Cnx1E)
SUOX is complete when MoCo and Heme are part of the enzyme. Heme synthesis requires:
- P5P (vitamin B6)
- Iron
- Zinc
- Radical SAM (Fe-S cluster dependent. SAM is made from methionine. Methionine salvage is dependent on B12, folate, and/or betaine. The enzyme that makes SAM, MAT, is inhibited by hydroxyl radicals that occur during oxidative stress – look into Bob Miller and Dr. Jill’s remedy using molecular hydrogen if you have high methionine levels of plasma amino acid indicating a block at MAT.)
- Riboflavin (vitamin B2) for FAD
Additional references….
kidney disease and sulfite retention…
https://pubmed.ncbi.nlm.nih.gov/10770971/
https://pubmed.ncbi.nlm.nih.gov/40058144/
https://pubmed.ncbi.nlm.nih.gov/14717909/
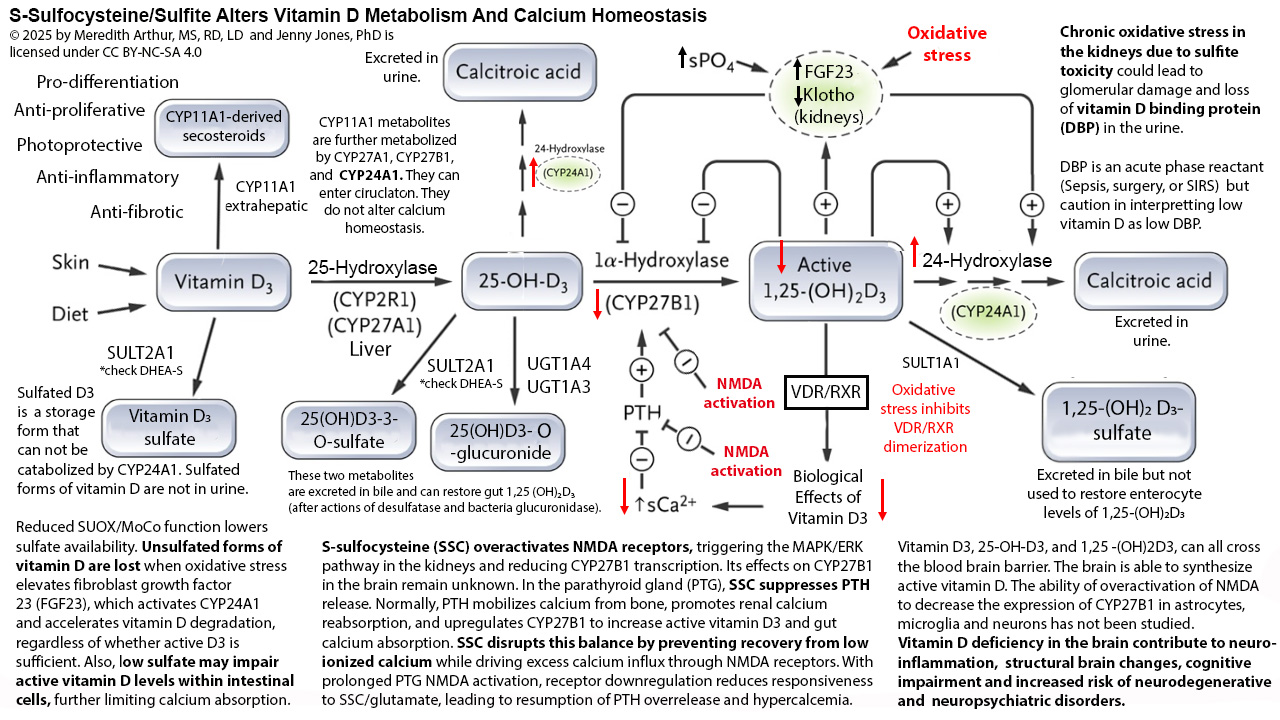
Adapted diagram for sulfite oxidase deficiency is from….
https://pmc.ncbi.nlm.nih.gov/articles/PMC9607355/
The cysteine and methionine restriction on the diagram are specifically for those with SUOX/MoCo deficiency. They are life saving measures, but unless you have this genetic disease, prolonged restriction of methionine or cysteine will only lead to worsening of your health (in my opinion, consult with your own provider). A methionine and cysteine restriction to the level needed for the genetic diseases of SUOX is not life sustaining.
Article on molybdenum exposure and chronic kidney disease https://www.sciencedirect.com/…/pii/S0147651324004767
Neutrophilic Contribution to Sulfite Toxicity © 2025 by Meredith Arthur, MS, RD, LD and Jenny Jones, PhD is licensed under CC BY-NC-ND 4.0![]()
![]()
![]()
![]()